Covid-19 Protocol: This Judgment was handed down remotely by circulation to the parties’ representatives by email and release to Bailii. The date for hand-down is deemed to be 7 April 2022.
Introduction. 3
The issues. 3
The witnesses and the skilled team.. 4
Some irrelevant matters. 5
Applicable legal principles - plausibility. 6
T 939/92 Agrevo/Triazoles. 6
Warner-Lambert v Generics [2018] UKSC 56. 10
Fibrogen v Akebia [2021] EWCA Civ 1279. 17
Identifying what it means to “work”. 25
T 0016/18 Sumitomo. 27
No requirement to file data. 28
No particular level of activity required. 28
Agreed common general knowledge. 28
Disputed common general knowledge. 29
Was a nanomolar Ki/IC50 necessary for therapy?. 29
Selectivity. 30
Predictability of in vitro and in vivo characteristics from structure alone. 31
Binding pockets of factor Xa; their relevance to inhibitor design. 31
Specific series of compounds. 33
The teaching of `652. 36
The claims of the Patent 44
Proposed amended claims. 45
Evidence and arguments on plausibility. 46
Plausibility of factor Xa binding. 46
The teaching on page 170. 47
3g quantity of apixaban. 47
The compounds synthesised; apixaban as a “typical” one. 48
Structural analysis. 51
P1 4-methoxyphenyl 53
The lactam at the P4 position. 54
The rigidified core. 54
Dr Redshaw’s evidence. 55
The differences in aggregate. 55
Conclusion on the structure case. 55
Plausibility of factor Xa binding - overall assessment 55
Plausibility of therapy. 56
Selectivity. 56
Clear and easy tests. 56
Non-therapeutic uses. 57
Standard or reference compounds. 57
Diagnostic assays. 57
Anti-coagulants. 58
Lead compounds. 58
Conclusion on non-therapeutic uses. 59
Obviousness over `131. 59
Obviousness of compound per se claims: Teva’s point 60
The proposed amendments. 61
Added matter 62
Clarity. 62
Conclusions. 62
Mr Justice Meade:
1. This is the trial of two actions in which the Claimants, respectively Sandoz Limited (“Sandoz”) and Teva Pharmaceutical Industries Limited (“Teva”) seek the revocation of European Patent (UK) 1 427 415 B1 (“the Patent”) in the name of Bristol-Myers Squibb Holdings Ireland Unlimited Company (“BMS”). The priority date is 21 September 2001.
2. The claims of the Patent relate to a compound called apixaban, sold by BMS under the name ELIQUIS and which is used for thromboembolic disorders.
3. There is also a corresponding SPC (SPC/GB11/042) but nothing separate turns on that: it is invalid if the Patent is invalid.
4. BMS has counterclaimed for infringement. Infringement is admitted by Sandoz and Teva in the event that the Patent is valid. So in substance this was a patent revocation trial.
5. Sandoz and Teva are separately represented but have made common cause, submitting joint skeleton arguments and sharing expert witnesses, and with their Counsel splitting the oral advocacy at trial. They have run the same arguments with one exception, an obviousness attack made by Teva alone. I will refer to Sandoz and Teva together as “the Claimants”.
6. Apixaban’s use in therapy depends on its activity as a factor Xa inhibitor. It is not in dispute that in fact apixaban has proven to be a potent factor Xa inhibitor and a useful therapeutic, but the central attack on the Patent is that it did not make plausible that apixaban would have any useful factor Xa inhibitory activity, or be useful in therapy, or for any other purpose.
7. It was common ground that the issue of plausibility should be tested by reference to the application for the Patent, published as WO 2003/026652 A1 (“`652”), because if plausibility had to be based on something that was only in the Patent and not in `652, there would be added matter. On that basis, an added matter squeeze fell away.
8. BMS has also applied to amend the Patent’s claims.
9. There has been related litigation in Canada, where BMS has been successful. But BMS did not submit that it could rely on the result there in this litigation and from what I have seen a very different legal standard applies there. Dr Camp, BMS’s medicinal chemistry expert, was involved in the Canadian litigation and that forms part of the background to his evidence in this case.
10. The issues were:
10.1 Some issues over common general knowledge (“CGK”). Much more was agreed than was in dispute.
10.2 Lack of plausibility. Lack of plausibility is not a ground for revocation in itself and it was run both as Agrevo obviousness and insufficiency. Neither side said it made a difference which head applies, and I agree in the light of the case law to which I refer below, in particular Warner-Lambert and Fibrogen. So I will just refer to lack of plausibility.
10.3 Obviousness over WO 00/39131 “Nitrogen Containing Heterobicycles as Factor Xa Inhibitors” (“`131”). This was not a “classical” obviousness attack: the Claimants do not say it was positively obvious to get to apixaban specifically from `131. Rather, they said that `131 contains very similar teaching to `652 about broad classes of compounds that include apixaban, and that there is no technical contribution in `652 over what `131 discloses.
10.4 (Teva only) That claims 1-6 exceed the technical contribution of the Patent and that in particular the claims to products per se are invalid even if some usefulness were to be plausible.
10.5 Allowability of the amendments, where the points were:
10.5.1 Whether the amendments were capable of curing any invalidity.
10.5.2 Lack of clarity.
10.5.3 Added matter.
11. The parties each called three experts. They were in:
11.1 Medicinal chemistry;
11.2 “The coagulation cascade”, which refers to the pharmacology/biochemistry of relevance;
11.3 Pharmacokinetics or DMPK (“drug metabolism and pharmacokinetics”).
12. Permission for the third of those, DMPK, was pressed for by BMS at the directions stage because it intended to argue that there was a contribution in relation to pharmacokinetics in `652. That argument collapsed with the oral evidence and I think the Claimants were right all along that the Patent is nothing to do with DMPK issues. There was no material dispute over the skilled team by the end of trial in any event: it would be a drug design/development team comprising (i) a medicinal chemist and (ii) a biochemist or pharmacologist, who would have relevant experience in industry. I have paid some minimal attention to evidence from the DMPK witnesses below, but it could easily have been given by one of the other experts and does not change my view on the skilled team.
13. There was no fact evidence.
14. The Claimants’ experts were:
14.1 Dr Sally Redshaw (medicinal chemistry);
14.2 Dr Robert Leadley (pharmacology/biochemistry);
14.3 Prof Kevin Read (DMPK).
15. BMS’s experts were:
15.1 Dr Nicholas Camp (medicinal chemistry);
15.2 Prof James Morrissey (pharmacology/biochemistry);
15.3 Dr David Taft (DMPK).
16. The only witness of whom personal criticism was made was Dr Taft. The Claimants submitted that his evidence was not credible, to such an extent that he must have been insincere in his defence of it. I reject any personal criticism of Dr Taft. The position he supported was indeed extreme and if it had not been given up on by BMS I would have rejected it, but I think he honestly believed it.
17. As to the other witnesses, points of detail were made about their approach and about their specific experiences. I will deal with those where they arise and to the extent I think them important, which they generally were not. My assessment of these specific points takes place against the backdrop of my more general view that the witnesses were well qualified and gave their evidence fairly and honestly.
18. A basic divergence between the parties was that the factor Xa knowledge was spread differently among their witnesses. For the Claimants, the factor Xa knowledge was possessed by Dr Leadley, with Dr Redshaw having no previous experience in relation to it. For BMS, Dr Camp had a lot of contemporaneous involvement relevant to factor Xa inhibition and Prof Morrissey had none of any significance. Although this probably meant that the Claimants’ witnesses together represented the notional skilled team better, in that medicinal chemists often have no direct experience of the specific target of a given project, I did not think it mattered.
19. Both sides raised matters which I think are irrelevant and which I will mention now so as to dismiss them.
20. For its part, BMS emphasised that apixaban has proved to be a very important and widely used drug by virtue of being a potent and selective factor Xa inhibitor. Indeed its closing written submissions said that this was the “central” issue. BMS also relied on the researchers behind apixaban having been awarded the “Heroes in Chemistry Award” from the American Chemical Society.
21. I think those matters are irrelevant. I have to assess plausibility on the basis of `652, the relevant specification for these purposes. Later findings about apixaban do not enter the picture. As to the award referred to, I am sure that it was merited, but I am equally sure that it was not given just for the work in `652 and that the standards applied were not those of patent law under the EPC. As Birss J, as he then was, said in Evalve v Edwards [2020] EWHC 514 (Pat) at [49], no judge wants to revoke a patent for a breakthrough, but this sort of evidence is really only introduced for prejudicial effect.
22. The Claimants on their side asserted that there was a practice among big innovators at around the priority date of deliberately leaving out data from patent applications filed for compounds per se in order to achieve broad protection without giving away their commercial intentions or technical information useful to their competitors. They said that either that practice had led to the omission from `652 of testing data about apixaban or the applicant had not done any such testing.
23. I think these assertions are also irrelevant and in addition I am not in any position to assess either whether there was such a practice, or what data existed that was not put into `652, or the reasons for any omission. My task is to assess whether what is in `652 renders plausible the qualities and uses of apixaban relied on.
24. As I have already said, lack of plausibility is not itself a ground of revocation, but arises under article 56 (inventive step) and article 83 (sufficiency) of the EPC, as carried into the Patents Act 1977 as conditions for grant and as reasons for revocation under s. 72.
25. I was addressed on a number of cases relating to the legal principles applicable in relation to plausibility. I do not think I need to deal with them all, but I will deal with the three central ones individually, and then with some others under the sub-themes to which they relate.
26. Agrevo is a seminal case in relation to the way plausibility arises in the law of obviousness under article 56 of the EPC, and its interaction with the contribution made by a patent.
27. Briefly, the patent under consideration claimed a class of triazole sulphonamides described by a Markush group. The claims were to the compounds per se and not use-limited, but the specification asserted usefulness as herbicides. The Examining Division held that the skilled reader would not expect all the claimed compounds would or could have that activity.
28. It was against this background that the TBA came to consider inventive step. In a very well-known passage it said:
2.4 During the oral proceedings the appellant argued that the only question arising under Article 56 EPC in the present case was whether or not, in the light of the above state of the art, a skilled person would have prepared, or tried to prepare, the claimed compounds of formula I (see point IV above), wherein R3 was optionally substituted phenyl. Article 56 did not expressly require, so he submitted, that the subject-matter of a patent application had to solve a technical problem, and that, accordingly, the issue of inventive step had to be decided without regard to the solution of any technical problem.
2.4.1 While the Board agrees with the appellant that the above question is the one which has to be answered under Article 56 EPC, it does not agree with his inference that the existence of a technical problem and its solution, including the problem of proposing alternatives to known activities (for example, chemical processes) or physical entities (for example, chemical compounds), is irrelevant to answering this question and so deciding the issue.
2.4.2 The reason for this is that it has for long been a generally accepted legal principle that the extent of the patent monopoly should correspond to and be justified by the technical contribution to the art (see T409/91, OJ EPO 1994, No. 3.3 and 3.4 of the Reasons, and T435/91, OJ EPO 1995, 188 , Reasons No. 2.2.1 and 2.2.2). Now, whereas in both the above decisions this general legal principle was applied in relation to the extent of the patent protection that was justified by reference to the requirements of Articles 83 and 84 EPC, the same legal principle also governs the decision that is required to be made under Article 56 EPC, for everything falling within a valid claim has to be inventive. If this is not the case, the claim must be amended so as to exclude the obvious subject-matter in order to justify the monopoly.
Moreover, in the Board's judgment, it follows from this same legal principle that the answer to the question what a skilled person would have done in the light of the state of the art depends in large measure on the technical result he had set out to achieve. In other words, the notional 'person skilled in the art' is not to be assumed to seek to perform a particular act without some concrete technical reason: he must, rather, be assumed to act not out of idle curiosity but with some specific technical purpose in mind.
2.4.3 For this reason, the Boards of Appeal consistently decide the issue of obviousness on the basis of an objective assessment of the technical results achieved by the claimed subject-matter, compared with the results obtained according to the state of the art. It is then assumed that the inventor did in fact seek to achieve these results and, therefore, these results are taken to be the basis for defining the technical problem (or, in other words, the objective) of the claimed invention (which problem may, as already stated above, be to provide a further—or alternative—process or physical entity, here a group of chemical compounds). The next step is then to decide whether the state of the art suggested the claimed solution of this technical problem in the way proposed by the patent in suit (see for example, T24/81, OJ EPO 1983, 133 , No. 4 of the Reasons). If the state of the art consists of written disclosures, it is often convenient, for practical reasons (see T439/92 —3.2.4 of 16 May 1994, No. 6.2.1 of the Reasons), to base this examination on one document which is most closely related to the claimed subject-matter as starting point, and to consider whether the other documents suggest to obtain the technical results which distinguish the claimed subject-matter from this 'closest state of the art'.
…
2.5 Using the above approach of the Boards, and having regard to the cited state of the art, in this case the Board considers that, if the claimed compounds were to be assumed not to have any technically useful property, then it could be postulated that the technical problem which is solved by the claimed compounds (or, in other words, the technical result achieved by them, on the basis of which the question of inventive step has to be decided), would be the minimalist one in such a situation, namely the mere provision of further (or alternative) chemical compounds as such, regardless of their likely useful properties.
2.5.1 Although the Board is not convinced that, in the absence of any technically useful properties, the claimed compounds could be regarded as being a technical invention at all (see Decision T22/82, OJ EPO 1982, 341 , No. 6 of the Reasons, where it was held that a chemical compound was not patentable merely because it potentially enriched chemistry, and that structural originality had no intrinsic value or significance for the assessment of inventive step as long as it did not manifest itself in a valuable property in the widest sense, an effect or an increase in the potency of an effect), the Board has nevertheless examined whether the notional person skilled in the art would have considered the claimed compounds as a solution of such a hypothetical 'technical problem'.
2.5.2 In this context, the appellant submitted that the skilled person would have faced thousands of possibilities of solving this problem, since even on the basis of known starting compounds and known synthetic methods, a practically unlimited number of chemical compounds would have had to be considered, and that a particular selection from this unlimited number of possibilities should be regarded as inventive, even if it was arbitrary, unless there was a direct pointer to the preparation of just these very compounds in the state of the art.
2.5.3 This argument, must, however, fail, since in the Board's judgment the answer to the question as to what a person skilled in the art would have done depends on the result he wished to obtain, as explained in point 2.4.2 above.
If this result is only to be seen in obtaining further chemical compounds, then all known chemical compounds are equally suitable as the starting point for structural modification, and no inventive skill needs to be exercised in selecting, for instance, the compound of formula XIV of D3 for this purpose. Consequently, all structurally similar chemical compounds, irrespective of their number, that a skilled person would expect, in the light of the cited prior art, to be capable of being synthesised are equally suitable candidates for solving such a hypothetical 'technical problem', and would therefore all be equally 'suggested' to the skilled person. It follows from these considerations that a mere arbitrary choice from this host of possible solutions of such a 'technical problem' cannot involve an inventive step (see also, for example, T220/84 of 18 March 1986, No. 7 of the Reasons). In other words, the Board holds that, in view of the underlying general legal principle set out in point 2.4.2 above, the selection of such compounds, in order to be patentable, must not be arbitrary but must be justified by a hitherto unknown technical effect which is caused by those structural features which distinguish the claimed compounds from the numerous other compounds. This consideration is also in line with a number of previous decisions of the Boards of Appeal of the EPO, such as, for example, Decision T01/80 ( OJ EPO 1981, 206 , No. 6 to 8 of the Reasons). In Case T119/82 (OJEPO 1984, 217), where in considering the argument that a person skilled in the art would neither consider nor propose an alternative process for preparing a known product which is 'exotic' or even disadvantageous, the deciding Board reached a similar conclusion, holding that a chemical process was not obvious only when the skilled person would have seen all its advantages, but also when he could clearly see its disadvantages or would not expect any improvement, provided that his assessment of the totality of the consequences was indeed correct (see Reasons, No. 16).
2.5.4 It follows directly from these considerations that a technical effect which justifies the selection of the claimed compounds must be one which can be fairly assumed to be produced by substantially all the selected compounds (see also, for example, T131/87 of 7 September 1989, No. 8 of the Reasons, T742/89 of 2 November 1992, No. 7.4 of the Reasons, T626/90 of 2 December 1993, No. 4.3.2 of the Reasons, and T741/91 of 22 September 1992, No. 4.2 and 4.3 of the Reasons).
2.6 Therefore, the Board holds that, contrary to the appellant's submission, the assessment of the technical contribution to the art must take account of the actual technical reason for providing the very compounds now being claimed, as distinct from the host of other theoretically possible modified chemical compounds. In this respect, the description (see page 3, lines 1 and 2) asserts that all claimed compounds do have herbicidal activity. Herbicidally active chemical compounds which are structurally similar to the claimed ones, since they are also triazole derivatives, are known from D3, D7 and D8 (see point 2.3.1 and 2.3.2 above). Any one of these documents may therefore serve as the 'closest state of the art' in the present case.
In view of this state of the art the technical problem which the present patent application asserts to solve is the provision of further (alternative) chemical compounds with herbicidal activity.
However, in the light of the Board's finding in point 2.4.3 above, this technical problem could only be taken into account if it could be accepted as having been solved, that is, if, in deciding the issue under Article 56 EPC , it would be credible that substantially all claimed compounds possessed this activity (see also point 2.5.4 above). Accordingly, the Board has examined whether this requirement is fulfilled.
29. Thus the key reasoning was that in relation to compounds for which utility was not credible (we would now say plausible), the only technical contribution is providing other compounds, and that cannot be inventive. In an ordinary sense one might say there was no reason to make any particular such other compound, but against the background of the non-solution of any problem there is a minimal reason which is good enough. I think the decision can be summed up by the sentence in 2.5.3 that “the selection of such compounds must not be arbitrary but must be justified by a hitherto unknown technical effect which is caused by those structural features which distinguish the claimed compounds …”.
30. This decision of the Supreme Court was the subject of extensive submissions by both sides and is central to my analysis.
31. The key claim in question was a second medical use claim, to the use of pregabalin for making a medicament for the treatment of neuropathic pain. The trial judge and the Court of Appeal had held it sufficient in the face of an attack that the specification did not make that use plausible. By a majority on this point (Lord Sumption, Lord Reed DP, Lord Briggs), the Supreme Court allowed the appeal on validity and decided that the claim was insufficient.
32. In dealing with this issue, Lord Sumption summarised the “patent bargain” at [17]:
17. Elementary as it is, it is worth reminding oneself at the outset of the juridical basis on which patents are granted, sometimes called the “patent bargain”. The inventor obtains a monopoly in return for disclosing the invention and dedicating it to the public for use after the monopoly has expired. The point was succinctly made by Lord Mansfield in Liardet v Johnson (1778), quoted in Hulme, “On the History of Patent Law”, (1902) 18 LQR 280, 285:
‘The condition of giving encouragement is this: that you must specify upon record your invention in such a way as shall teach an artist, when your term is out, to make it - and to make it as well by your directions: for then at the end of the term, the public shall have benefit of it. The inventor has the benefit during the term, and the public have the benefit after ...’
The principle remains the foundation of modern patent law, and is recognised in the case law of both the United Kingdom and the European Patent Office. In EXXON/Fuel Oils (T 409/91) [1994] OJ EPO 653, at paras 3.3 and 3.4, the EPO Technical Board of Appeal observed that it was-
‘the general legal principle that the extent of the patent monopoly, as defined by the claims should correspond to the technical contribution to the article in order for it to be supported, or justified. … This means that the definitions in the claims should essentially correspond to the scope of the invention as disclosed in the description. … Although the requirements of articles 83 and 84 are directed to different parts of the patent application, since article 83 relates to the disclosure of the invention, whilst article 84 deals with the definition of the invention by the claims, the underlying purpose of the requirement of support by the description, insofar as its substantive aspect is concerned, and of the requirement of sufficient disclosure is the same, namely to ensure that the patent monopoly should be justified by the actual technical contribution to the art.’
The principal conditions of validity, novelty, inventive step, industrial application and sufficiency are all, in one way or another, directed to satisfying the principle thus expressed.
33. Following a long analysis of the statutory provisions and key EPO and UK decisions, some of which I will return to below, Lord Sumption summarised at [35] - [37]:
35. All of these judgments deal with highly fact-specific issues arising from objections or potential objections on the ground of insufficiency. When reading them, it is important not to miss the wood for the trees. The fundamental principle which they illustrate is that the patentee cannot claim a monopoly of a new use for an existing compound unless he not only makes but discloses a contribution to the art. None of them casts doubt on the proposition that the disclosure in the patent must demonstrate in the light of the common general knowledge at the priority date that the claimed therapeutic effect is plausible. On the contrary, they affirm it: see Allergan at paras 26, 37, and Bristol at para 3.2.
36. The Court of Appeal’s statement of the effect of the plausibility test has already been quoted (para 20 above). They considered that the threshold was not only low, but that the test could be satisfied by a “prediction … based on the slimmest of evidence” or one based on material which was “manifestly incomplete”. Consistently with that approach, they considered (paras 40, 130) that the Board’s observations in Salk laid down no general principle. I respectfully disagree. The principle is that the specification must disclose some reason for supposing that the implied assertion of efficacy in the claim is true. Plausibility is not a distinct condition of validity with a life of its own, but a standard against which that must be demonstrated. Its adoption is a mitigation of the principle in favour of patentability. It reflects the practical difficulty of demonstrating therapeutic efficacy to any higher standard at the stage when the patent application must in practice be made. The test is relatively undemanding. But it cannot be deprived of all meaning or reduced, as Floyd LJ’s statement does, to little more than a test of good faith. Indeed, if the threshold were as low as he suggests, it would be unlikely to serve even the limited purpose that he assigns to it of barring speculative or armchair claims.
37. Plausibility is not a term of art, and its content is inevitably influenced by the legal context. In the present context, the following points should be made. First, the proposition that a product is efficacious for the treatment of a given condition must be plausible. Second, it is not made plausible by a bare assertion to that effect, and the disclosure of a mere possibility that it will work is no better than a bare assertion. As Lord Hoffmann observed in Conor Medsystems Inc v Angiotech Pharmaceuticals Inc [2008] RPC 28, para 28, “it is hard to see how the notion that something is worth trying or might have some effect can be described as an invention in respect of which anyone would be entitled to a monopoly”. But, third, the claimed therapeutic effect may well be rendered plausible by a specification showing that something was worth trying for a reason, ie not just because there was an abstract possibility that it would work but because reasonable scientific grounds were disclosed for expecting that it might well work. The disclosure of those grounds marks the difference between a speculation and a contribution to the art. This is in substance what the Technical Board of Appeal has held in the context of article 56, when addressing the sufficiency of disclosure made in support of claims extending beyond the teaching of the patent. In my opinion, there is no reason to apply a lower standard of plausibility when the sufficiency of disclosure arises in the context of EPC articles 83 and 84 and their analogues in section 14 of the Patents Act. In both contexts, the test has the same purpose. Fourth, although the disclosure need not definitively prove the assertion that the product works for the designated purpose, there must be something that would cause the skilled person to think that there was a reasonable prospect that the assertion would prove to be true. Fifth, that reasonable prospect must be based on what the TBA in Salk (para 9) called “a direct effect on a metabolic mechanism specifically involved in the disease, this mechanism being either known from the prior art or demonstrated in the patent per se.” Sixth, in Salk, this point was made in the context of experimental data. But the effect on the disease process need not necessarily be demonstrated by experimental data. It can be demonstrated by a priori reasoning. For example, and it is no more than an example, the specification may point to some property of the product which would lead the skilled person to expect that it might well produce the claimed therapeutic effect; or to some unifying principle that relates the product or the proposed use to something else which would suggest as much to the skilled person. Seventh, sufficiency is a characteristic of the disclosure, and these matters must appear from the patent. The disclosure may be supplemented or explained by the common general knowledge of the skilled person. But it is not enough that the patentee can prove that the product can reasonably be expected to work in the designated use, if the skilled person would not derive this from the teaching of the patent.
34. I also think [40] is important context to later parts of Lord Sumption’s judgment in the light of Counsel for BMS’s submissions:
40. Warner-Lambert’s second argument is that the courts below were wrong to reject later published data as relevant. This submission also is contrary to the legal basis of this particular head of insufficiency. We know that pregabalin works for the treatment of both peripheral and central neuropathic pain, because like any other medicament on the market, it underwent demanding clinical trials after the priority date, the results of which were made public. On that basis it received marketing authorisation for all neuropathic pain. This is always the case for a commercially valuable medicament, and no other kind will be worth litigating about. The question is not whether it works but whether the contribution to the art consisting in the discovery that it can be expected to work has been sufficiently disclosed in the patent. The inherent difficulty of demonstrating this before clinical trials is taken into account in the modest standard (ie plausibility) which is applied to test it. This point was made by the EPO Technical Board of Appeal in Salk, at para 8:
‘Sufficiency of disclosure must be satisfied at the effective date of the patent, ie on the basis of the information in the patent application together with the common general knowledge then available to the skilled person. Acknowledging sufficiency of disclosure on the basis of relevant technical information produced only after this date would lead to granting a patent for a technical teaching which was achieved, and, thus, for an invention which was made, at a date later than the effective date of the patent. The general principle that the extent of monopoly conferred by a patent should correspond to, and be justified by, the technical contribution to the art, has to be kept in mind.’
This does not mean that subsequent data is never admissible in a dispute about sufficiency, but the purpose for which it is admitted is strictly limited. Where the asserted therapeutic effect is plausible in the light of the disclosure in the patent, subsequent data may sometimes be admissible either to confirm that or else to refute a challenger’s contention that it does not actually work: see, for example, AstraZeneca/Omeprazole Na (T 1677/11) (27 November 2012, unpublished), Merck, Sharp & Dohme/Pharmaceutical nanoparticulate composition of a Tachykinin receptor antagonist (T 0210/11) (17 July 2014, unpublished). But it cannot be a substitute for sufficient disclosure in the specification. As the EPO Technical Board of Appeal observed in Johns Hopkins University School Of Medicine/Growth differentiation factor-9 (T 1329/04) [2006] EPOR 8 at para 12, (cited above), it cannot be a substitute for sufficient disclosure in the specification.
35. The argument before me also focused on [53]-[54]:
53. Floyd LJ said (para 133) that he was “fortified” in his conclusions by a further consideration, which the judge had not relied on, namely that
‘… it was established through the evidence that the skilled team would be encouraged by the data in the patent to carry out simple tests (which are themselves identified in the patent) to confirm the suitability of pregabalin for peripheral neuropathic pain. I would have thought, on the basis of that evidence (as I think the judge did) that the specification had thereby made a contribution to the art which would justify a claim to peripheral neuropathic pain.’
The “simple tests” that Floyd LJ was referring to were the Bennett and the Kim and Chung tests for peripheral neuropathic pain; and the evidence that he had in mind was that of Dr Scadding, the expert clinician called by Actavis and Mylan: see paras 119-120 and 127. Dr Scadding had accepted that “the skilled person would be encouraged by the data in the patent to ask the neuroscientist to test pregabalin for neuropathic pain.” Professor Wood, the expert neuroscientist called by Actavis and Mylan who would notionally have been asked to carry out these tests, gave more guarded answers when he was asked to deal with the point in cross-examination: Day 2, pp 265-269. His evidence, in summary, was that there were “no data whatever about neuropathic pain in the patent”, but that he would be encouraged by the broad terms of the claims to try many tests, including the Bennett and the Kim and Chung tests. There were, he said, “many different pain mechanisms that can give apparently similar symptoms”, for which there were different models, and it would be necessary to test for all of them. Some were difficult to test for. It was put to him that even the Bennett and the Kim and Chung tests would not provide definitive proof of efficacy, because it was a “step by step process”. His final answers on this point fairly reflect the tenor of his evidence, so far as one can judge from the transcript:
‘A. … So one would just carry out an analysis of all these different models, to see where the drug had better utility than present medication.
Q. The data in the patent would give you sufficient motivation to carry out further tests and step-by-step you would reach the stage where you have demonstrated that pregabalin was effective for the treatment of pain?
A. It would certainly inspire you to analyse its activity in a broad range of pain models. Of course, this would be useful for the clinician attempting to exploit the drug in treating various different types of human pain. Animal models are not ideal, but they are always a useful pointer for the clinician.
Q. A useful starting point?
A. Absolutely.’
I am conscious of the danger of an appellate court analysing extracts from a transcript of evidence on complex and inter-related technical questions, where so much depends on the impression that the witness’s evidence as a whole has made on the trial judge. But in the absence of any discussion of this point by the judge, I feel unable to attach the same importance to it as Floyd LJ did. There is, however, a more fundamental objection to it, which is well brought out by the evidence which I have cited from Professor Wood. In classical insufficiency cases, where the question is whether the disclosure in the patent enables the skilled person to perform the invention, the skilled person may be assumed to supplement the disclosure by carrying out simple tests. In cases like this one, where the invention is novel but the objection of insufficiency is that the claim exceeds the disclosed contribution to the art, the role of hypothetical “simple tests” is necessarily more limited. As the EPO Technical Board of Appeal observed in Johns Hopkins, at para 12, the specification can be said to contribute to the art if it solves a problem, but not if it merely poses one. Or as Lord Hoffmann observed in a passage that I have already quoted, the notion that something is “worth trying” cannot be enough without more to justify a monopoly. The specification in the present case says nothing about neuropathic pain of any kind. It says nothing about central sensitisation, which is said to provide a link between neuropathic and inflammatory pain. The mere fact that the skilled team, faced with an apparent discrepancy between the breadth of the claims and the absence of supporting data in the specification, would be encouraged to fill the gap by carrying out tests of its own, serves only to confirm the absence of any disclosed contribution to the art.
54. I conclude that Claim 3 of the patent and the other claims relating to neuropathic pain were invalid for insufficiency. The disclosure did not contribute any knowledge of the art capable of justifying a claim to a monopoly of the manufacture of pregabalin for the treatment of neuropathic pain of any kind.
36. In opening, Counsel for the Claimants stressed the principle from [17] (and [25]) that the patent monopoly must correspond to and be justified by the technical contribution to the art, and the second and seventh points from Lord Sumption’s summary: that bare assertion of an effect does not provide plausibility and that matters supporting sufficiency must appear from the patent’s specification, albeit that it may be supported by the common general knowledge.
37. Sensing that BMS might be arguing that Warner-Lambert is confined to second medical use claims, Counsel for the Claimants pointed out in closing that while that was the context of the case, Lord Sumption’s analysis of plausibility was not limited to it, and that a number of key cases that he considered (Agrevo at [23], Johns Hopkins at [24], and BMS/Dasatinib also at [24]) were not about second medical use patents. I agree with this but I do not think that BMS took such a stance in the end, in any event.
38. For its part, BMS stressed in opening the relatively low standard for plausibility identified by Lord Sumption at [37], third point: “not just … an abstract possibility that it would work but because reasonable scientific grounds were disclosed for expecting that it might well work”.
39. In closing, BMS sharpened its argument and developed a more detailed analysis of Warner-Lambert in connection with the significance of a patent specification not containing efficacy/activity data.
40. The first part of this contention was that there is no requirement as such that a patent must contain efficacy data because plausibility can be established by a theory, in particular a theory based on the structure of a compound (or class of compounds). I agree with this, and in itself I do not think the Claimants disputed it. When I come to the facts I will therefore have to assess whether there is a theoretical basis for the plausibility of apixaban arising from structure.
41. The second part of the contention was that Lord Sumption had left open the possibility that tests not done by the patentee but which might be done by the reader of a specification, could be relevant to plausibility. This submission turned on Lord Sumption’s statement in [53] that in the sort of case where insufficiency is said to arise from exceeding the technical contribution, the “role of hypothetical ‘simple’ tests is necessarily more limited”.
42. Counsel for BMS argued that this meant that although Lord Sumption thought the Court of Appeal had gone too far in its reliance on the possibility of doing the Bennett and Chung tests once “encouraged” by the specification, such tests could have a role. The purpose of this submission was to create a legal basis for the argument that the reader of `652 would see something of potential value by working out what the patentee was likely to have done and, encouraged by that but having no data, would themselves test apixaban.
43. I disagree with BMS’s argument on this point. Lord Sumption clearly rejected the encouragement-plus-later-tests argument in [53], and all that he meant by simple tests having a more limited role was a reference back to the case law of the EPO on post-filed data that he had identified in [40]. That he was rejecting a role for tests which had not been done for inclusion in a patent specification is clear from [53] itself in his reference to Johns Hopkins to the effect that setting a problem is not a contribution and that the notion of “worth trying” does not without more justify a monopoly.
44. BMS sought to reinforce its argument on this front by reference to BMS/Dasatinib T0488/16, at 4.6.2. BMS argued that that case showed that one of the factors that the TBA considered in assessing plausibility was the availability of tests. In fact, what the Board referred to was the lack of availability of any CGK tests for verifying the assertion in question, and its statement was that that “further aggravated” the lack of plausibility arising from the specification. In complete isolation from any context I can see how BMS might argue that it could be inferred that tests could theoretically have a role, but in reality that is plainly not what the Board was saying. I note that BMS/Dasatinib was referred to by Lord Sumption at [24] and although he referred to a different paragraph in the decision (4.9) he was dealing with the issue of post-filed data, so this too is a reason to reject BMS’s reliance on the decision.
45. In my view my analysis of plausibility should be firmly guided by the points in [37] of Warner-Lambert and by the principle laid out by that case that a contribution by the patentee that is in the specification is needed. The latter is important because, as I hope will become clear when I address the facts, in very large measure, if not entirely, BMS’s case for plausibility arises not from anything in `652 but from matters which it contends were CGK. CGK is not BMS’s contribution.
46. In Fibrogen, the claim in question was as follows (quoted at [14]):
A Use of a heterocyclic carboxamide compound selected from the group consisting of
B pyridine carboxamides, quinoline carboxamides, isoquinoline carboxamides, cinnoline carboxamides, and beta-carboline carboxamides
C that inhibits hypoxia inducible factor (HIF) prolyl hydroxylase enzyme activity
D in the manufacture of a medicament for
E increasing endogenous erythropoietin
F in the prevention, pretreatment, or treatment of anemia associated with kidney disease,
G wherein the anemia is associated with chronic kidney disease.
47. In this Swiss claim, feature B defines an extremely wide group of compounds, feature C requires that they inhibit a certain enzymatic activity, and feature E requires that the medicament manufactured must have the effect of increasing endogenous erythropoietin. The clinical conditions to be addressed are identified in features F and G.
48. The trial Judge (Arnold LJ, sitting at first instance) held, as summarised by the Court of Appeal at [37]-[40], that the claim was insufficient because of the extreme breadth of feature B: the skilled person would have thought that the specification was promising that substantially all compounds with that structure would have the relevant therapeutic efficacy, but that was not plausible, and testing across the enormous scope of feature B to identify all the compounds covered would be an undue burden.
49. The Court of Appeal disagreed. The key part of its analysis for my purposes are at [49]-[59] - this quote is rather long but I do not think anything would be gained by my paraphrasing it:
The law - insufficiency
49. To grapple with this, I start with the legislation. The 1977 Act provides that to be valid the specification must disclose the invention "clearly enough and completely enough for it to be performed by a person skilled in the art". This corresponds to Art 83 EPC although the Act uses the word "performed" instead of the Convention's phrase "carried out", but there is no difference. Everything else is judge-made law, working out how this principle applies in different sets of circumstances. As the judgment does in paragraph [347] it is useful to see that this single ground can be classified into three types of objection - classical insufficiency, Biogen insufficiency aka excessive claim breadth, and uncertainty. Nevertheless one does need to take care not to read too much into brief summaries of what those categories amount to and not to treat them like statutes.
50. Just as the kinds of insufficiency can be put into categories, so too can the kinds of case to which they apply. The issue in this case is about alleged excessive claim breadth as it applies to inventions which are concerned with compounds and classes of compounds whose utility is in some kind of medical therapy.
51. The most up to date general statement of the relevant law of insufficiency, particularly as it relates to claim breadth in this context, is that made by Kitchin LJ in Regeneron v Genentech in the Court of Appeal at paragraphs [95] to [103]. The whole passage repays careful reading. It is not necessary to set it all out. The fourth principle of the six which Kitchin LJ identifies relates to inventions defined in general terms and the requirement of a reasonable prediction:
'If the invention discloses a principle capable of general application, the claims may be in correspondingly general terms. The patentee need not show that he has proved its application in every individual instance. On the other hand, if the claims include a number of discrete methods or products, the patentee must enable the invention to be performed in respect of each of them.
Thus if the patent has hit upon a new product which has a beneficial effect but cannot demonstrate that there is a common principle by which that effect will be shared by other products of the same class, he will be entitled to a patent for that product but not for the class, even though some may subsequently turn out to have the same beneficial effect: see May & Baker Ltd v Boots Pure Drug Co. Ltd. (1950) 67 R.P.C. 23, 50 . On the other hand, if he has disclosed a beneficial property which is common to the class, he will be entitled to a patent for all products of that class (assuming them to be new) even though he has not himself made more than one or two of them.'
‘112. In my opinion there is nothing difficult or mysterious about [a principle of general application]. It simply means an element of the claim which is stated in general terms. Such a claim is sufficiently enabled if one can reasonably expect the invention to work with anything which falls within the general term. For example, in Genentech I/Polypeptide expression (T 292/85) [1989] O.J. EPO 275 , the patentee claimed in general terms a plasmid suitable for transforming a bacterial host which included an expression control sequence to enable the expression of exogenous DNA as a recoverable polypeptide. The patentee had obviously not tried the invention on every plasmid, every bacterial host or every sequence of exogenous DNA. But the Technical Board of Appeal found that the invention was fully enabled because it could reasonably be expected to work with any of them.
113. This is an example of an invention of striking breadth and originality. But the notion of a 'principle of general application' applies to any element of the claim, however humble, which is stated in general terms. A reference to a requirement of 'connecting means' is enabled if the invention can reasonably be expected to work with any means of connection. The patentee does not have to have experimented with all of them."
100. It must therefore be possible to make a reasonable prediction the invention will work with substantially everything falling within the scope of the claim or, put another way, the assertion that the invention will work across the scope of the claim must be plausible or credible. The products and methods within the claim are then tied together by a unifying characteristic or a common principle. If it is possible to make such a prediction then it cannot be said the claim is insufficient simply because the patentee has not demonstrated the invention works in every case.
101. On the other hand, if it is not possible to make such a prediction or if it is shown the prediction is wrong and the invention does not work with substantially all the products or methods falling within the scope of the claim then the scope of the monopoly will exceed the technical contribution the patentee has made to the art and the claim will be insufficient. It may also be invalid for obviousness, there being no invention in simply providing a class of products or methods which have no technically useful properties or purpose.”
52. It may be a matter of taste only but I prefer to refer to this fourth principle as reasonable prediction rather than simply plausibility, however whatever it is called, it is the same principle.
53. To apply the reasonable prediction principle one has to take three steps. First one must identify what it is which falls within the scope of the claimed class. Second one must determine what it means to say that the invention works. In other words what is it for? Once you know those two things, the third step can be taken: to answer the question whether it is possible to make a reasonable prediction the invention will work with substantially everything falling within the scope of the claim.
54. In a paradigm case of a Swiss style claim to the use of a class of compounds defined in a Markush formula to treat a disease, the first two steps are simple and the question will be whether it is possible to make a reasonable prediction that substantially all the molecules within the Markush class will work to treat the disease. In terms of functional and structural limitations in claims, in this simple case the structural limitation defines the class and is considered at the first step and the functional limitation defines the therapeutic effect and is addressed at the second step. The significance of the existence of inactive compounds within the Markush formula will be a matter of fact and degree but the fact they exist does not matter if it does not falsify the reasonableness of the prediction. Also and similarly the fact that active compounds within the formula turn out to be unsuitable as clinically approved agents for reasons unrelated to efficacy itself, such as side effects profiles, bioavailability and the like, is also unlikely to falsify the reasonableness of the prediction, depending again on this being a matter of degree. These issues will also play a role in analysis of any undue burden.
55. However in other cases the first step also involves a separate functional limitation too, in addition to the use to treat a disease. Claims with such double functional features are not so unusual. Twenty years ago the crucial claim in Lilly ICOS v Pfizer [2000] EWHC Pat 49) was to the use of a cGMP PDE enzyme inhibitor for the treatment of male erectile dysfunction. There was no structural limitation in that claim at all. The claim in Regeneron v Genentech is another example. Although there was a debate before us about how to characterise that claim, essentially it was a claim to the use of a product defined at least partially in functional terms for use in treating certain non- cancerous diseases characterised by excessive blood vessel growth. The functional definition of the products claimed was that they had to be antagonists to human vascular endothelial growth factor (VEGF). Amongst other things the court below in that case had held that it was possible to make a reasonable prediction that VEGF antagonism could be used to treat all the relevant diseases, and on appeal the Court of Appeal rejected the insufficiency attack holding at [134] that "The judge had ample evidence before him upon which to conclude that it was plausible that VEGF antagonism could be used to treat any non-neoplastic neovascular disease.
56. Thus Regeneron is an example of the three step test I have referred to applied to a claim with double functional features. To distinguish between these two kinds of functional feature I will refer to "step one functional features" (such as VEGF antagonism) and "step two functional features" (such as treating the relevant diseases). It will be a matter of construction to work out what sort of functional features one is dealing with.
57. In some cases the second step is the aspect which is a bit more involved. So in Idenix v Gilead, claim 1 was to a Markush class of molecules (see Kitchin LJ para [61]). The claim language did not include any reference to what they were for and so one could not answer the question at the second step by looking at the words of the claim. This is also not unusual. If the compounds are new, then a claim to those compounds will be novel without including a claim feature which refers to what they are actually for. However that does not prevent the reasonable prediction principle being applied. In fact the answer in Idenix was clear from the patent specification. That showed that the point of the invention was to treat infections caused by viruses in the Flaviviridae family. So one can assess the validity of the claim on the basis that it is a claim to compounds with anti-Flaviviridae activity, which is what Kitchin LJ said at paragraphs [113] and [124]. So, in the language coined above, anti-Flaviviridae activity was a step two functional feature. The issue in Idenix arose in the context of inventive step but the same approach applies to reasonable prediction/plausibility. Note that this does not mean that claims to compounds per se are actually limited to using the compounds for treating Flaviviridae infections, but for the purposes of assessing questions like inventive step and reasonable prediction/plausibility, one needs to know what the compounds are supposed to be useful for. In fact in Idenix the outcome of the third step was against the patentee. The court held that it was not plausible that substantially all the claimed molecules would be effective against Flaviviridae infections, and hence it was Agrevo obvious and also insufficient for lack of plausibility for the same reason (see paragraphs [129] and [140]).
58. Before leaving this it is worth expanding briefly on Agrevo. If one was performing a Pozzoli analysis of inventive step in such a case, the inventive concept would be the compounds for treating Flaviviridae infections. In the EPO, one would ask what the problem to be solved is, and the answer would be the same - to treat Flaviviridae infections. Just as in Agrevo itself, so in Idenix , the claim was to a Markush class of compounds with no limitation to the use they were for, but that did not prevent the tribunal from determining what they were for by reading the patent specification. In Agrevo itself the use was as herbicides. So the EPO's problem/solution approach would ask the question whether the claimed molecules were or were not obvious to use as herbicides. They may well not have been. However Agrevo is authority for the proposition that there is a prior question. Before one can investigate inventive step that way, the tribunal must be satisfied that the alleged problem to be solved is indeed solved by the claimed subject matter. The Agrevo question is whether it is credible or plausible that the claimed compounds have the alleged beneficial property. If they do then that useful property can be employed to formulate the problem to be solved. If they do not then the claim lacks inventive step because drawing up a list of compounds with no plausible utility is not an act of invention. As Regeneron v Genentech makes clear in the passage cited above, the Agrevo question is the same as the question whether it is possible to make a reasonable prediction that the invention will work with substantially everything falling within the scope of the claim.
59. I turn to the third step in reasonable prediction. The solidity of the basis for a given prediction, or putting it another way, the degree of plausibility required, was something addressed by Lord Sumption in the Supreme Court in Warner Lambert v Generics. As far as I know nothing turns on that aspect of this issue in the present case.
50. The Court went on to apply these, and their central reason for disagreeing with the trial Judge was that the claim did not extend to all the compounds of feature B, but only to those which also had the functional features C and E.
51. In my view it is important, especially in the light of the proposed amendments to the Patent, which are intended to assimilate its claims more closely to those in Fibrogen, for me to have regard to the three steps in [53]. But they pose very different tasks for me in this case compared to those which arose in Fibrogen.
52. Step 1 was the most difficult in Fibrogen, but before me there is no problem: the claims cover only apixaban.
53. Step 2, what it means to say that the invention works, was considered by the Court of Appeal at [56]-[58] by reference to “step one functional features” (which there was the enzyme inhibition) and “step two functional features” (which there was the treatment effect on anaemia). The Court of Appeal identified that even where the claim in question is a compound claim and does not recite functional features, it should be possible to deal with step 2 by reading the specification as, the Court pointed out, had been done in Idenix v Gilead [2016] EWCA Civ 1089 and Agrevo itself. This is useful guidance which I will follow when I consider the facts of the present case but the dispute before me about what it means that the invention “works” is more complex than that which the Court of Appeal had to consider in Fibrogen.
54. Step 3 was touched on by the Court of Appeal at [59] by reference to Warner-Lambert. Nothing turned on step 3 in the Court of Appeal’s decision because there was no dispute that if the patentee was right about the earlier steps, it should prevail on step 3. So I do not think there is anything in Fibrogen to affect the application of the principles from Warner-Lambert that I have identified above.
55. BMS also relied on Fibrogen in relation to the role of tests for a functional effect when it comes to establishing plausibility. This was based on [95]-[97]:
95. Having run through the EPO decisions, I conclude as follows. There is clear support for a test based on the narrow reading of the extract from paragraph 6.6.9. The principle based on the narrow reading would not be contentious. Also, if the facts are like those in the Bayer/Reach through case then a question along the lines of paragraph [366] may arise. However the only decision which supports the principle of law as it is stated in the judgment at [366] is T 544/12 Princeton/OLED itself. That is not a sufficient basis to reach such a radical conclusion. In my judgment paragraph [366] is wrong. The right test is as follows. If one has a claim with a functional feature which defines the claimed compounds, or a mix of such structural and functional features, it must be possible, without undue burden, both to identify compounds which satisfy the relevant test, and to find out whether any given compound satisfies the test. However it is not necessary as a matter of law, for sufficiency (or for Agrevo ), simply because a claim contains functional features (or a mix of functional and structural features) to establish that the skilled person can identify all or substantially all the compounds which satisfy the test.
96. Finally, if the law does not require the identification of substantially all such compounds, the question remains, how many is enough? Take the facts of the present case. The claims like claim 8A with structural and functional language at step one clearly claim a wider class than the particular compounds C, E, F, J and K identified in the patent as likely to have therapeutic efficacy. Even if one adds on the 100 or so compounds identified in the patent at paragraphs [0072]–[0077], the claim is plainly intended to be much wider than that too. In terms of a promise, the wider claim is a promise or assertion that there are more useful compounds within the class than the ones identified by name in the patent. Bearing in mind the ultimate issue is all about breadth of claim, in such a case the question is how many is enough?
97. I believe the answer is in two parts. For claims of this type, it must be possible for the skilled person, without undue burden, to identify some compounds beyond those named in the patent, which are within the claimed class and therefore are likely to have therapeutic efficacy. Otherwise the contribution is no more than the named compounds and the wider claim is too wide and unsupported by the disclosure. Second and separately, it must also be possible for the skilled person to work substantially anywhere within the whole claim ( Kymab is one example, in which inventive step was needed to be able to work in a part of the claim which was not otherwise available to the skilled team from the specification, and another is the non-functional 2'-methyl-up-2'-fluoro- down sub-class of the Markush formula in Idenix). So it must be possible for the skilled person, given any sensible compound within the structural class (or substantially any), to apply the tests without undue burden and work out if it is a claimed compound.
56. I reject BMS’s argument on this point. I agree with the Claimants that Birss LJ at that stage was dealing with how to determine the scope of the claim. He was clearly not at that stage dealing with the question of use of later experiments at step 3, because on step 3 he had simply referred to Warner-Lambert.
57. Fibrogen represents important recent guidance from the Court of Appeal on sufficiency/plausibility and I intend to bear in mind the three steps identified above. But it is important to recognise too that the context was very different from the present case. The patentee had made an invention which was the identification of the role of a biological mechanism (the enzymatic inhibition) which enabled a treatment effect. This was an important principle. The present case is quite different: BMS asserts that its invention is in identifying a single compound which, based on a known mechanism and known structural understanding would treat a recognised kind of condition of concern. So there really is no useful analogy to the step one and two functional features of [56] in Fibrogen.
58. In passing, I note Birss LJ’s statement in [54] that the fact that active compounds turn out to have problems unrelated to efficacy itself, such as side effects and bioavailability, will not usually lead to a lack of reasonable prediction/plausibility. I consider that it is relevant to the selectivity issue.
59. As I say, the arguments in the case before me on this issue were more complex than those which arose in Fibrogen. In particular, the arguments before me covered the issues of how to address a situation where the specification makes multiple statements of utility, and how to address the situation where the specification makes an assertion of a technical advance which turns out to be overstated.
60. The Claimants relied on Pharmacia v. Merck [2001] EWCA Civ 1610. That was a classical insufficiency case about compounds useful as anti-inflammatories. Two potential effects of the claimed compounds were under consideration: their ability to have an anti-inflammatory effect and their ability to be “Cox II selective” which would imply that they did not cause gastric problems. The claims were claims to classes of compounds as such and did not recite any particular use.
61. The defendants had done experiments to prove that compounds within the classes claimed were inactive as anti-inflammatories and lacked Cox II selectivity. One of the patentee’s arguments was that it did not matter if the compounds were not Cox II selective as long as they were active anti-inflammatories. The patentee also argued that the invention of a compound claim was the compound as such and that activity was not required.
62. Aldous LJ, with whom Sedley LJ and Arden LJ agreed (the latter giving some additional concurring reasons), roundly rejected the notion that there could be a meaningful invention just in identifying new compounds without any use (see e.g. [61]), and of course that is consistent with Agrevo, Warner-Lambert, Fibrogen and other cases in this jurisdiction and in the EPO. He also held, at [20] and [26] in particular, that based on construing the specification the skilled reader would have identified the invention as the provision of compounds which were both anti-inflammatory and gastric-sparing by reason of Cox II selectivity.
63. BMS responded by pointing to cases where obviousness was in issue where the Court has been open to the notion that a patentee can rely on a more modest level of technical contribution than that identified in the specification. In particular, it cited the remarks of Floyd LJ in Generics v Yeda [2013] EWCA Civ 925 at [63]-[65]:
63. The problem and solution approach to obviousness requires the court or tribunal to judge inventiveness by reference to what it is that the invention brings with it: its technical effect or advance. Like any other fact relevant to an issue, however, it must be open to being refuted. In doing so one is not judging the obviousness of the claimed invention by reference to later evidence: one is simply defining by evidence what it is that the invention is or brings with it.
64. The rule in John Hopkins that a technical effect relied upon must be made plausible by the specification, and cannot be established for the first time by subsequent evidence, was not in issue before the judge and is not in issue in this appeal, and I need say no more about it. It is sufficient to say that it does not provide a basis for the different rule arrived at by the judge as to whether subsequent evidence may be used to negate an effect made plausible by the specification. I respectfully disagree with the judge when he concluded that it was not open to Mylan to challenge an effect made plausible by the specification. For my part, I cannot see any principled objection to the admission of evidence as to the true nature of the advance made by the invention in connection with an objection of lack of inventive step.
65. The mere fact that the primary technical contribution relied upon by the patentee is negated by evidence does not of course lead inexorably to the conclusion that the patent is obvious. The patentee may advance an alternative less ambitious technical contribution of the kind discussed in AgrEvo. The party attacking the patent will still have to persuade the court that that invention was obvious, and do so by reference to what the skilled team would have known and done at the priority date. In the present case, however, Mr Waugh was content to put his case on the basis of the inventive contribution propounded in the patent. He did not, for example, argue in the alternative that the invention simply provided further compounds of the same activity as copolymer-1.
64. It also referred to the well-established approach of the EPO under which, including for the purposes of assessing plausibility, the technical advance provided by a patent can be reformulated in the light of the closest prior art, which may have been unknown to the patentee when writing the specification. As an example it cited T 0116/18 Sumitomo at 13.7.2, and it pointed out that the patentee can in such circumstances rely even on effects not mentioned in the specification, so long as they are from the same field of use and do not change the “character of the invention” and preserve the “spirit of the original statement of the invention”.
65. I record that BMS submitted that Pharmacia was implicitly overruled by Generics v. Lundbeck [2009] UKHL 12, but I did not understand the argument and it was effectively withdrawn during oral closing submissions. More realistically, it submitted that Pharmacia did not contain an analysis of Agrevo obviousness (which is substantively correct, although Arden LJ did cite it at [164] in the course of her rejection of the patentee’s argument that merely identifying new chemicals was enough). BMS also pointed out that in Eli Lilly v HGS [2012] EWCA Civ 118 at [31] (this was the judgment of the Court of Appeal following the Supreme Court’s decision), Jacob LJ had said that Pharmacia would be reasoned differently then, with regard to Agrevo.
66. In my view BMS is right overall on this point, and in cases where the objection is of lack of plausibility in an Agrevo-type situation, a patentee is not necessarily limited to the most demanding teaching of utility in the specification and is entitled to try to rely on a less ambitious degree of utility, or a utility of a different but related kind. I think this is logical because the Agrevo-type of objection is that there is no technical contribution at all and a patentee ought to be able to meet it by showing that there is some contribution even if it turns out that the contribution is less than the patentee thought, perhaps because of some new prior art. I agree with BMS that to decide otherwise would severely cut across the EPO approach as identified by the Court of Appeal in Generics v Yeda.
67. I do not think that this approach is necessarily inconsistent with Pharmacia, which was not about plausibility but about whether compounds in the broad class claimed actually had the qualities taught by the specification. Furthermore, the point did not matter in Pharmacia because the patent there was invalid for many reasons, and was insufficient even if only anti-inflammatory activity was required.
68. So I conclude that what it means for the invention to “work” is to be determined from the specification where the claim is not explicit (I do not think this in itself was in dispute), but that the patentee is not restricted to the most ambitious assertion made. In some cases the patentee may be able to rely on a more limited contribution, but this must be fact-dependent and will still have to find a basis in the specification.
69. I record that the Claimants accepted that where a specification teaches multiple independent utilities for new compounds a patentee may be able to meet an allegation of lack of technical contribution/plausibility by making good only one of them but said that in the present case the teaching in the specification was cumulative; that the non-therapeutic applications were premised on apixaban having the necessary qualities for a therapeutic and meeting further requirements.
70. I have already touched on this TBA decision above in relation to the ability of a patentee to reformulate its technical contribution. More importantly, it was in this decision that the TBA referred important questions about plausibility to the Enlarged Board of Appeal of the EPO.
71. The decision is a careful and detailed review of the position in the EPO as regards plausibility. It identifies the divergent lines of authority in the case law of the TBA, and refers to what it calls the “Ab initio plausibility” line of case law (see 13.4), represented in particular by Johns Hopkins T 1329/04 and Salk T 609/02, and the “Ab initio implausibility” line of case law (see 13.5). It identifies the Supreme Court’s decision in Warner-Lambert (at 13.5.5) as being in accordance with the “Ab initio plausibility” standard, and I agree with that. It also more briefly identifies a third line of case law, “No plausibility” (at 13.6).
72. The result of the reference to the Enlarged Board of Appeal in this case will be extremely important for the EPO and for all EPC member states, but (other than as an example of reformulating the technical contribution) the TBA’s decision in itself does not affect my analysis in this case because I am bound by Warner-Lambert.
73. BMS argued that there is no requirement to file data in an application in order to establish plausibility; that an effect may be made credible from the structure of a compound, for example. It relied on T 0184/16 Galencium Health by way of example. I do not believe the Claimants disputed this, and I accept it. The Claimants instead met BMS’s structural arguments on the facts.
74. BMS argued that no particular level of biological or therapeutic activity is required by law when it comes to plausibility. It based this submission on [14] in Eli Lilly v. HGS (supra). I do not believe the Court of Appeal was making any such general statement in that case.
75. The reason for BMS’s making this point was to lay the ground for a submission that any level of factor Xa activity would be good enough, even if it could not achieve anything of known utility, because it could serve as a reference point. So, BMS would say, even if `652 only rendered it plausible that apixaban had trivially low factor Xa activity, it could serve as a comparator for better compounds.
76. While recognising that patent specifications do not have to reach a standard of excellence or perfection, and a “working prototype” will often be good enough, there comes a point where activity loses any practical meaning and I think this argument goes beyond that point. In my view the law requires a technical contribution of some, even if low, real significance. There is no contribution in disclosing a uselessly low degree of activity so that comparisons can be made with something which is useful. BMS’s argument on this point is not really different from the sort of nihilistic argument that novel compounds with no known use can be put into service as ballast, or the like.
77. In accordance with recent practice in the Patents Court, the parties agreed and provided a document setting out agreed CGK. Usually I edit these down for inclusion in my judgments, excluding, if there is much of it, material which has turned out to be of no relevance to the issues and/or of no use in assisting the reader to understand the technical content of the judgment.
78. In the present case, I do not think that is the best approach, mainly because there is relatively little superfluous material, and I therefore attach the whole agreed CGK document as Annex A, which I incorporate by reference. I invite readers of this judgment to read it and then resume here, but for the benefit of those already familiar with the basics of proteins, enzymes, enzyme inhibitors, and drug discovery/medicinal chemistry, they may wish to pass quickly through the respective sections in paragraphs 77 to 121. Also, (i) although I have not deleted it, the material about DMPK issues from paragraph 133 to the end of the document is not of any remaining relevance sufficient to justify spending any significant time on it, and (ii) the “N.B.” before paragraph 109 can be ignored.
79. The parties also prepared a document indicating the disputes about CGK. At my invitation they updated this following trial to indicate disputes which had fallen away. Helpfully, they included evidence references to the matters still in dispute, for which I am grateful. I have used the document as a checklist and source of references in preparing this section of my judgment. In some instances the Claimants said that issues were not relevant but BMS asked that I decide them; I have decided them.
80. I will deal with the issues in the order they appeared in the parties’ list, although I have not used their precise wordings.
81. The Claimants asserted that the skilled person would consider that for a factor Xa inhibitor to be potentially useful in treating thromboembolic disorders it would need Ki/IC50 values in the nanomolar range (and materially less than 10 µM or 1 µM).
82. This was a potentially important issue because `652, in its key passages, only asserts that among the compounds studied some had been found to have a Ki of less than 10 µM. So the Claimants argued that even if that teaching concerned or covered apixaban (which they dispute), it does not disclose a useful level of activity.
83. On this issue, I conclude that the Claimants were correct. Dr Leadley gave clear evidence in support of the Claimants’ position which he maintained under cross-examination; Dr Redshaw also supported it, although I thought this was more a matter for the pharmacologist. There was also a sound theoretical basis for such a high level of activity being needed (which was that the inhibitors have to act on factor Xa in the prothrombinase complex), and contemporary literature supporting it.
84. Prof Morrissey’s initial position in his written evidence (footnote 6 to his first report) was consistent with this, and when he was taken to the literature he generally accepted the Claimants’ position. When challenged that 1-10 µM was too high to be useful clinically, he responded that there were some lead compounds in that range, but “lead” compound in this context means as a starting point for research, not as a clinically useful result in itself, as he accepted.
85. In its written closing submissions, BMS submitted that there was no fixed level of potency in the field because no factor Xa inhibitor drug had ever made it into the clinic. While that is true, and there was no precise level of potency known to be needed, this submission does not meet the point that a nanomolar potency was the order thought necessary, and that 1-10 µM was known as a matter of CGK not to be good enough. BMS also argued that part of the drive for nanomolar potency was the need to compete with other compounds, but even if that was part of the picture, my assessment based on the expert evidence and literature to which I have referred just now is that there were solid, objective reasons for requiring such potency for therapy. Finally, BMS submitted that less good potency might be offset by other positive qualities such as good bioavailability, but this was a speculative argument: there was no evidence that the art accepted that that sort of trade off could make up for a potency in the 1-10 µM range or that there was a perception that the undoubted work going on to look for nanomolar potencies might not be necessary.
86. On the basis of the evidence of Dr Redshaw and Dr Leadley, the Claimants argued that it was CGK that selectivity over other serine proteases (including trypsin, thrombin, aPC, plasmin, and tPA) was a necessary feature of a potentially useful factor Xa inhibitor, with a high degree of selectivity for factor Xa being necessary to avoid off-target effects that could result in lack of efficacy or side-effects. They said the necessary level of selectivity was at least 100-fold over any other serine protease.
87. The considerations applicable to the other serine proteases would be understood by the skilled person to differ. In particular, trypsin is important in the gut and for an oral drug activity against it would be problematic (but this problem would not arise with parenteral administration). For other serine proteases mentioned above the issue would be seen as being not just this kind of side-effect but also a potential problem with efficacy, because those other serine proteases are involved in the coagulation cascade.
88. Prof Morrissey’s oral evidence supported this. For example, he said that with a factor Xa inhibitor for use to prevent clotting in a tube, inhibition of trypsin would be irrelevant, but for treating thromboembolic diseases effectively selectivity against other serine proteases would be very important.
89. Contemporaneous literature also stressed the desirability of selectivity, as Prof Morrissey agreed when it was put to him. Dr Camp agreed that the standard panel of serine proteases tested for by Lilly in this field at the time included plasmin, tPA, urokinase, aPC, thrombin, trypsin and others.
90. The Claimants also pointed out that `652 at page 171 says that “compounds of the present invention may also be useful as inhibitors of serine proteases, notably human thrombin, Factor VIIa, Factor IXa, Factor XIa, urokinase, plasma kallikrein, and plasmin”. I do not think that has relevance to the state of the CGK although it might to the issue of plausibility, in which context I take it into account below. `652 also refers on the same page to the possibility of thrombin inhibition being useful to treat other conditions.
91. BMS did not really dispute in closing that selectivity was known as a matter of CGK to be something that should be considered. It argued that Dr Redshaw had given only limited evidence about likely problems with thrombin and trypsin, but that is to overlook the evidence of Dr Leadley (to which Dr Redshaw had cross-referred) and the cross-examination of Prof Morrissey.
92. I conclude that it was CGK that selectivity against other serine proteases was important and was tested for, and that it was CGK that cross-reactivity could lead to side effects, or loss of activity, or both.
93. However, this does not mean that it was CGK that until selectivity of this kind was demonstrated the skilled team would think that a compound which had good activity against factor Xa was necessarily not even a plausible therapeutic. They would think lack of selectivity was a risk that would need testing for in due course.
94. While grateful to the parties for their efforts on the CGK, I found this a poorly-characterised dispute. I am not sure that it was about CGK at all, really.
95. It is the business of medicinal chemists to relate structure to activity; they do make predictions based on structure and on knowledge of existing compounds’ characteristics, but their confidence in those predictions varies greatly. I agree with BMS’s observations in their written closing argument, where they contrasted Dr Redshaw’s statement that a “prediction” as to factor Xa inhibition based on structure alone was impossible with Dr Camp’s statement that an “educated assessment” (about apixaban) could be made. BMS said that the statements were not necessarily inconsistent, because for Dr Redshaw a “prediction” might simply imply a high degree of confidence whereas for Dr Camp an “educated assessment” would imply a lower one.
96. In my view it was CGK that structure could be a useful pointer in relation to activity but its importance was extremely context-dependent. How useful structure is in the present case has to be looked at with all the facts, and I do that below.
97. Again, I am afraid I found it rather hard to understand what the parties disagreed about and why. BMS said that I should decide the extent to which the binding pockets of factor Xa were CGK and the Claimants said no finding was necessary.
98. Paragraphs 123 and 124 of the agreed CGK document seem to me to deal with this issue. In this particular instance I will quote from that document. Those paragraphs say the following:
Factor Xa structure
123. The crystal structure of human factor Xa had been published in 1993. Subsequently, crystal structures with bound inhibitors (including DX-9065a - see below) were also published, showing how synthetic small molecule inhibitors bind to the factor Xa binding pockets. [Redshaw 1/59; Camp 1/6.53 & 6.74]
124. Proteases have at their active sites a number of specificity pockets (S1, S2, etc.) into which the substrate binds. The crystal structures of factor Xa bound to different inhibitors showed that the key binding pockets for small molecules binding to factor Xa are S1 and S4, which were well characterised. S1 is a deep, narrow pocket with hydrophobic walls and an aspartic acid (Asp189) at its base. The S4 pocket has distinct sub-regions; a ‘hydrophobic box’ and a negatively charged cation binding hole. [Redshaw 1/59; Camp 1/6.59 & 6.60]
99. In addition, the evidence clearly established that the 2001 paper by Maignan and Mikol of Aventis Pharma “The Use of 3D Structural Data in the Design of Specific Factor Xa Inhibitors” was CGK.
100. Counsel for BMS put the following passages from Maignan and Mikol to Dr Redshaw:
100.1 From the Abstract:
In the case of fXa several structure based drug design strategies have been followed because of the difficulty in growing fXa co-crystals routinely. This has led to the use of surrogate proteins such as trypsin. Factor Xa inhibitors for which the binding mode has been determined experimentally or modeled are described in this review. The inhibitors are divided into three fragments: a P1 group, a central scaffold and a P4 group. In this review, interactions in each sub-site of fXa with various inhibitor fragments have been examined at the molecular level and were shown to bind, in most cases, independently of the rest of the molecule. Knowledge of the 3D structure of the binding mode of ligands to target proteins has been successfully applied in designing fXa inhibitors with enhanced specificity, affinity and has provided hints to modulate the physicochemical properties of the small molecule ligand.
100.2 From page 161, left hand column:
The knowledge of the structural data has been instrumental in designing selective compounds but it does not provide direct clues on how to improve pharmacokinetics parameters. However, it has successfully guided medicinal chemists in the selection of groups that could be introduced into inhibitors to modulate the physicochemical properties of the compounds without compromising potency and selectivity. This will be exemplified in the review.
100.3 From page 163, right hand column:
Each fXa inhibitor reviewed in this paper can be divided into three fragments: the P1 group which binds in the S1 pocket, a linker or a central scaffold designed to project the substituents appropriately into the pockets and the P4 group which interacts with the S4 pocket. Hereafter are described the interactions - modeled or observed experimentally - between the sub-sites of fXa (or a surrogate enzyme e.g. trypsin or thrombin) and the inhibitors. The analysis presented is made from the viewpoint of the binding pockets of the protein. This linked fragment approach where ligands are constructed from building blocks that show conserved binding modes, is the organizational logic for this review. Inhibitors can be generated using this fragment based approach, provided the orientation of each part of the inhibitors matches the one from the single building blocks. There are exceptions that will be discussed but the majority of cases are amenable to this approach. This has some bearings on the strategy for the development of fXa inhibitors and for other serine protease inhibitors.
101. She essentially accepted that these matters were CGK, and I hold that they were. They provide a general framework for thinking about the design of factor Xa inhibitors in a rational way by reference to the binding pockets, but stop well short of offering certainty.
102. I did not really understand the Claimants to dispute the CGK status of Maignan and Mikol and indeed they relied on it quite a lot. Nor did they say that knowledge of the S1 and S4 pockets was not CGK to some degree, since of course they appear in the agreed document. If their disagreement related to how confident a prediction could be made from this knowledge then I agree that there was not certainty. I think the real disagreement was what conclusions could be drawn in the detailed circumstances of apixaban and `652, but that is not a dispute about the CGK as such.
103. It is worth pointing out that paragraph 131 of the agreed CGK document identifies five factor Xa inhibitors as CGK:
103.1 RPR-120844 (Rhone Poulenc Rorer) [Leadley 1.5.86(c); Camp 1/6.76]
103.2 Betz compound 6 (DuPont) [Camp 1/6.77; Redshaw 1/Table 1, row 2]
103.3 ZK-807834 (Berlex Bioscience) [Leadley 1/5.86(a); Camp 1/6.78 - 6.80; Redshaw 1/Table 1, row 9]
103.4 DPC-423 (DuPont) [Leadley 1/5.86(d); Camp 1/6.84 - 6.86; Redshaw 1/Table 1, row 11]
103.5 RPR-208815 (Rhone Poulenc Rorer) [Leadley 1/5.86(c); Camp 1/6.83; Redshaw 1/Table 1, row 4]
104. I make reference to DPC-423, in particular, below. These compounds were agreed CGK in addition to a Daiichi compound called DX 9065a, one of the first published lead compounds, also described (more fully than the above) in the agreed CGK document at paragraphs 127-130.
105. However, there remained a dispute about whether three other series of compounds were CGK. They were:
105.1 The AstraZeneca series of factor Xa inhibitors which included the compound with the following structure [Camp 1/6.87 - 6.89; Redshaw 2/36 - 37]:
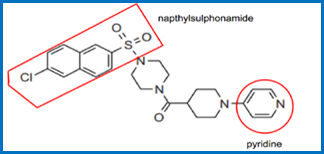
105.2 The Eli Lilly series of factor Xa inhibitors which included the compound with the following structure [Camp 1/6.90 - 6.94; Redshaw 2/39 - 45]:
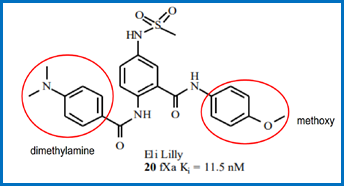
105.3 The DuPont series of factor Xa inhibitors which included the compound with the following structure [Camp 1/6.90 - 6.94; Redshaw 2/39 - 45]:
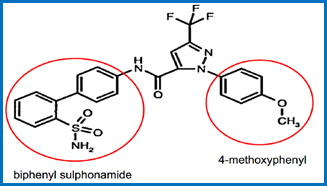
106. Of these, the Claimants said that no finding needs to be made about the first (AstraZeneca) but BMS invites me to decide whether it was CGK.
107. I will deal with the second and third, both of which relate to an issue concerning the move towards more neutral groups being used, especially at the P1 position, and then return to AstraZeneca.
108. The second series, from Eli Lilly, is identified in five review articles:
108.1 Zhu & Scarborough, 1999, “Recent Advances in inhibitors of factor Xa in the prothrombinase complex”;
108.2 Ries, 2000, “Factor Xa inhibitors - a review of the recent patent literature”;
108.3 Rai, 2001, “Perspectives on Factor Xa Inhibition”;
108.4 Betz, 2001 “Recent advances in Factor Xa inhibitors”;
108.5 Maignan and Mikol;
although in Zhu & Scarborough no data are given. Dr Redshaw said that the compounds in the series were not particularly notable for their activity and that no bioavailability data are given. Bioavailability was the reason she had not identified them in her own literature search and I agree with BMS and Dr Camp that in that respect she cast her net too narrowly; the ordinary medicinal chemist would not have used it as a basis for exclusion when seeking to gain a CGK appreciation of existing compounds. On the basis of these frequent references in the review literature and the fact that activity data are provided even if not exceptionally good, I hold that this Lilly series was CGK. Further support for that conclusion comes from the fact that the work on these Lilly compounds was part of a move towards neutral binders at the P1 position, and it was accepted that that general notion was part of the CGK.
109. The third series, from DuPont, is mentioned in two of the review articles (Zhu & Scarborough 1999, Betz 2001) but there are no data for it in them. Its lower degree of prominence and the complete lack of data satisfy me that it was not CGK. This seems to be of minimal, if any, importance however, because BMS made its case about neutral binders on the footing of the Lilly series and other matters, and its reliance on this DuPont series faded during oral closing submissions.
110. As to the AstraZeneca series, the specific compound depicted above was in clinical trials at the priority date. Dr Redshaw did not include it in her Table 1, which was of “Compounds of interest to the skilled medicinal chemist from the literature search” because she thought there were no in vivo efficacy data for it. Prior to her oral evidence she noticed that there were data, and corrected her report. Although a little grudging in the way she phrased it, she accepted in cross-examination in substance that it should have been included in Table 1, which amounted to an acceptance that the skilled medicinal chemist would have been aware of it from routine research necessary to understand the field. So I hold that it was CGK. But it was of minimal importance to the arguments at trial, forming just a very minor part of the picture in relation to the move to less basic structures and then neutral S1 binders; its binding mode was unknown.
111. It is worth articulating what I mean when I say that a compound (or series) was CGK in this context. I believe my understanding is also what the parties intended in their submissions. This was a field where the understanding of factor Xa inhibitors and their modes of binding and the dependency on structure was developing but incomplete. Work was building up by accretion and was reflected in the sort of review articles I have referred to above. But no compound had been approved as a drug. So for a compound to be CGK means that it was a widely known compound recognised to have a significant place in the developing knowledge in the field. It does not imply perfect understanding of the compound’s binding or that the compound was likely to make an active substance in a drug. This point has some significance in relation to, for example, BMS’s deployment of the Lilly series in relation to the 4-methoxyphenyl point on the argument for plausibility based on structure.
112. As I have said, it was common ground that plausibility should be assessed from `652, the application for the Patent, to avoid any issue of added matter.
113. `652 is over 400 pages long and it would be impractical and unhelpful to try to summarise it all. Neither side tried to do so and the arguments focused on certain specific aspects, as will I. I recognise and have borne in mind however that the relevant teaching has to be assessed as a whole.
114. The title of `652 is “Lactam-containing compounds and derivatives thereof as Factor Xa Inhibitors”. The field of invention (page 1 lines 7-12) is similarly described in a bit more detail, thus:

115. In the following “Background of the Invention” section, `131 is mentioned at page 2 line 25 to page 3 line 5 (although I think little turned on this):

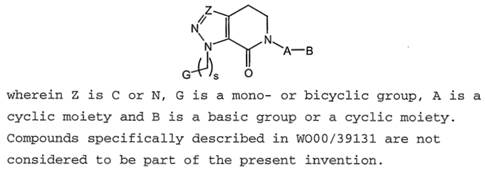
116. There is some general teaching about factor Xa starting at page 5 line 21:


117. This is however just the CGK.
118. There follows (page 6 lines 6-35 and especially the list (a) to (g)) a passage to which much importance was attached by BMS at the start of the trial and was the reason why it sought and obtained permission for a DMPK expert:

119. BMS’s case was that the matters (a) to (g) were in some way a disclosure relating to the beneficial qualities that had in fact been achieved by compounds of the invention generally and apixaban in particular. In my view however it is clear that (a) to (g) are just a generic checklist of things it would be desirable to achieve and have no relation to anything actually demonstrated. The fact, as shown in cross-examination of Dr Taft, that the same language is to be found in other patent applications emphasises and supports this conclusion but is not necessary to it. Although Dr Taft stoutly maintained his evidence, BMS gave up on this part of its case and I need say no more about it.
120. There follows a “Summary of the Invention” section starting at page 7. This begins with the statement (lines 2-5):

121. This maintains a general focus on lactams. The rest of page 7 down to line 32 contains a number of consistory statements, all of which focus on therapy, and they provide the context for the introduction of a broad class of compounds identified from page 7 line 33 to page 8 line 5:


122. The Claimants argued that down to this point the emphasis is all on therapy and that that conditions the “objects” that are said to be “achieved”. However, the text does refer to “[t]hese and other” objects and as will appear, non-therapeutic uses are also disclosed later on.
123. The specification then moves into a long section titled “Detailed Description of Preferred Embodiments” from page 8 line 7 onwards. Attention focused on embodiments 6, 7 and 8, and I will need to explain in more detail below where they fit into the arguments. For the moment I will briefly introduce them.
124. Embodiment 6 is too long to quote here, but includes numerous structures with options for substituents, some of which are lactams and some of which are not (the last 8 on page 67 are not).
125. Embodiment 7 is disclosed on page 67 to page 68:
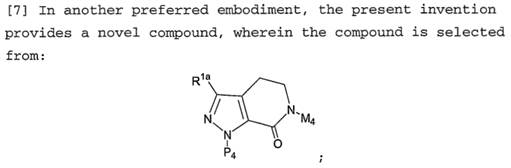
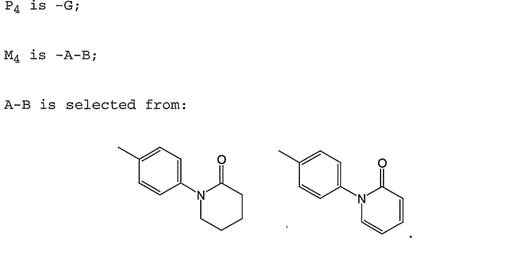
126. Embodiment 8 is a list of 74 individual compounds. Apixaban is the last on page 69:

127. Embodiment 15 from pages 117 to 119 lists another 124 compounds.
128. In his evidence, Dr Camp referred to these two lists as the First Group and the Second Group.
129. The Claimants emphasised the large number of compounds that fall within the embodiments defined by Markush formulae (of which there are more than just the ones I have specifically identified) and that embodiment 15, which is another list of specific compounds, does not include apixaban. These points are both true but not of much importance.
130. From pages 134 to 144 there is a Definitions section and from pages 143 to 168 a Synthesis section. Nothing turns on them.
131. Much more importantly for my purposes, from page 168 there is a section entitled “Utility”. This begins with the following statement:

132. There follows some general description of thromboembolic disorders and their causes, and then there is the following statement:
133. This is of potential significance to the Claimants’ argument about selectivity.
134. There follows a description of the chromogenic assay said to have been used at page 169 lines 22 to 34:

135. Experimental details and an explanation of the calculation of Ki are then given and which I need not quote, followed by a section on page 170 which was a key focus of the arguments before me:

136. In my view, the only statement of work actually done is that “a number of compounds” were tested and had a Ki of 10 µM or less. The statements about lower Kis for preferred/more preferred/still more preferred compounds are aspirational targets, and the statement that the utility of “the compounds of the present invention” was confirmed is an assertion that an inference can be drawn from the tests that were done. I understood that BMS accepted this.
137. I note that it seemed that Dr Camp had thought that the statements in relation to lower Kis, down to the nanomolar level, were also statements of fact of what had actually been achieved and this error coloured his evidence to some extent, which I have taken into account.
138. The Claimants accepted (based on Evans Medical’s Patent [1997] EWHC 359 Pat) that they cannot go behind the statement of fact as to what was done, but say that they can challenge the validity of the inference said to be based on it. I agree with this. I note that Dr Redshaw expressed doubts about whether anything at all had been tested in any way. This was a similar error to that made by Dr Camp (although in the opposite direction) and I have taken it into account, too.
139. I note that there is no indication in this text itself of which or how many compounds were tested or with what specific result, and there is no reference to apixaban. BMS accepted this but said that the whole picture of the disclosure of `652 must be considered, and at that general level I agree. So I must go on to consider the other later disclosure and the evidence before reaching any conclusion about this passage.
140. There follows, from the bottom of page 170 down to page 171 line 17, a description of a rabbit model for antithrombotic effect. Nothing really turns on this.
141. At page 171 lines 18-34 there is a discussion of the effect of the “compounds of the present invention” on other serine proteases, including thrombin, of the usefulness of such effect, and of tests having been done (but it is not said on which compounds):

142. Details of the assay follow, and there is a statement of results having been achieved at page 172 lines 17-21:

143. This parallels the report of factor Xa inhibitory activity in its level of detail.
144. So far the focus of `652 is on therapy, but non-therapeutic applications are referenced at page 179 line 17 to page 180 line 10:


145. From page 188 onwards a large number of examples are given. There are 140 numbered examples, of which apixaban is number 18. Synthesis and characterising data are described for 110.
146. A large number of other “representative examples” follow from page 298; these are just lists of compounds.
147. Claims mirroring the embodiments appear from page 316 onwards. BMS referred to claim 8 being to the compounds of the First Group and claim 15 being to the compounds of the Second Group. There are also claims to pharmaceutical compositions and uses.
148. Claim 1 of the Patent is as follows:
“1. A compound, which is represented by formula (1):
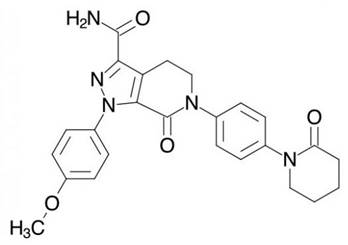
or a pharmaceutically acceptable salt thereof.”
This is apixaban.
149. Claim 2, not said to be independently valid, is as follows:
“2. A compound according to claim 1, which is represented by the formula (1).”
and I refer to it only because it is mentioned in claim 7, which is said to be independently valid:
“7. A compound of claim 1 and 2 for use in treating a thromboembolic disorder.”
150. The proposed amended claims are as follows (proposed additions underlined):
“7A. A compound of claim 1 or 2 that is a factor Xa inhibitor for use in treating a thromboembolic disorder.
7B. A compound of claim 1 or 2 that is an effective factor Xa inhibitor for use in treating a thromboembolic disorder.”
151. Counsel for BMS clarified that BMS would seek one or other of these and not both; he said the reason for putting forward both was in case there was a dispute about whether the word “effective” was necessary or appropriate.
152. BMS’s position was that the amendments were sought so that the amended claims corresponded to those in Fibrogen, with factor Xa inhibition as a step one functional feature and treatment of a thromboembolic disorder as a step two functional feature.
153. I will break plausibility down into:
153.1 Plausibility of factor Xa binding.
153.2 Plausibility of therapy.
153.3 Selectivity.
153.4 Non-therapeutic uses.
154. BMS’s case has multiple aspects to it:
154.1 Interpretation of the teaching on page 170 of `652.
154.2 Reliance on the 3g quantity of apixaban made.
154.3 An analysis of the compounds reported in `652 as having been synthesised to show that apixaban was a “typical” compound.
154.4 An analysis based on its structure that apixaban was likely to be an effective factor Xa inhibitor.
154.5 The availability of simple tests to determine the potency and selectivity of apixaban (and other compounds in the Patent) and the fact that they would show positive results. I think this falls into a category of its own and I deal with it separately.
155. BMS argued that these things taken together mean that `652 makes it plausible that apixaban is an effective factor Xa inhibitor. As I understood it, BMS’s case was that that (factor Xa inhibition in itself) was enough for claim 1, but it also relied on it being plausible on the basis of the above that apixaban was useful as a therapeutic for thromboembolic conditions and for the non-therapeutic purposes that I have mentioned above.
156. The Claimants disputed all aspects of BMS’s case. A particular focus of their submissions was that the question of plausibility must depend on what was disclosed about apixaban itself and not on “detective work” directed at inferring what data the patentee had or might have generated but not included in the specification. They also said that:
156.1 Even if the specification of `652 made it plausible that apixaban had been tested and found to have a Ki of the order of 10 µM, that was inadequate for therapeutic use;
156.2 That the non-therapeutic uses needed just as good a level of activity against factor Xa and so were also not plausible and/or were not sufficient in law.
157. Before assessing these arguments, I need to explain in more detail what BMS said about each.
158. Although its written submissions relied on the very general statements in e.g. the abstract of `652, in oral submissions Counsel for BMS accepted that they were not themselves good enough for plausibility and I agree with that, since at most they are bare assertions of utility. So the focus fell on the sentence at page 170 lines 28-32.
159. Counsel for BMS submitted that although it was not explicitly stated which compounds were tested, the skilled reader would assume that all the synthesised compounds, or at least the vast bulk of them, had been tested. The basis for this was said to be that `652 described the invention as being about lactams, that the patentee could only have tested compounds that were actually made, and that there was no point making them unless they were going to be tested.
160. Counsel for BMS did however accept that the skilled reader would infer that not all the compounds tested would have been successful; some might have failed. I agree with this.
161. In my view BMS seeks to read far too much into the sentence. On its own it would not be understood as standing with any reliability for anything more than it says, which is that some unidentified compounds had been tested with activities at the level indicated, and that utility for some broader class (i.e. broader than just the ones tested) could, in the patentee’s opinion, be inferred. What that broader class might be cannot be worked out, both because of the lack of detail and because of the inherent ambiguity in the expression “compounds of the present invention” in this sort of specification where many different Markush formulae are given.
162. Further, there is no way from this sentence alone to draw any sort of inference about any individual compound, be it apixaban or any other. There is simply no information, and given Counsel for BMS’s acceptance that some compounds might also have failed, there is no way for the reader to know of any particular compound whether it was good or bad.
163. To be fair, I do not think that BMS really seriously maintained a case that the disclosure on page 170 was enough on its own. It therefore sought to tie it to apixaban by means to which I will now turn.
164. As I have already said, apixaban is Example 18 in `652 and at page 222 line 25 it is identified that 3.07g was ultimately made. Although the Claimants raised some minor questions about the reporting of the quantities reported in the stages of the work I did not think they undermined the conclusion that of the order of 3g was made.
165. In addition, I find that that was the most of any compound reported to have been made in `652, by some distance.
166. There is no explicit disclosure of why the patentee made that amount. BMS said that the reader would infer that it was because early results had been favourable and the patentee wanted to take work on the compound forwards. The evidence of the DMPK experts (this was an isolated instance where their evidence was relevant) was that this was possible, with the further work intended being, possibly, second species pharmacokinetics or early toxicology.
167. The Claimants responded that there were other possible reasons, such as making apixaban as an intermediate on the way to making something else (although Dr Redshaw could not make any concrete suggestion) or as a thrombin inhibitor, which seems possible given the teaching of `652 on that topic, if not especially likely.
168. In cross-examination Dr Camp was taken to a 2003 publication by Scott Sheehan of Lilly (“A four component coupling strategy for the synthesis of D-phenylglycinamide-derived non-covalent factor Xa inhibitors”) where a similar large amount was made of a compound which was not successful. He accepted on the basis of it that the amount of a compound made could not be taken as an indicator of success in every case; one possibility was just that “the chemistry worked better”.
169. There was, Dr Camp accepted, no evidence in any of the CGK review articles of the authors selecting compounds for review or inclusion based on the amount made.
170. In her oral evidence, Dr Redshaw maintained her overall position that judgments could not be made about a compound’s qualities from the amounts made.
171. The 3g point is not completely without relevance. It is a point which, unlike other aspects of BMS’s case, is relatively free of hindsight, in the sense that it sets apixaban apart from the other exemplified compounds based on information in `652 itself that I think the skilled reader would notice.
172. However, in its substance it is a very weak point. Lacking any data, one does not know why the patentee made such a quantity and reasons other than factor Xa inhibitory activity are real possibilities. And I do not see how the point can go any further than that the patentee thought that apixaban was promising. A bare assertion to that effect in `652 (bare in the sense of lacking data or reasoning) would not have been any use in establishing plausibility, as is clear from the second point in [37] in Warner-Lambert. But `652 does not even contain such an assertion.
173. This point needs some explanation before its weight can be assessed.
174. Dr Camp undertook a detailed exercise in which he looked at the compounds listed in embodiments 8 and 15, having particular regard to those which had been synthesised. What he did was to convert the names of the compounds into structures, then worked out which were in one of the lists and which had been synthesised, and grouped them by core structure and by their functional groups. He looked at which features occurred the most often. The Claimants referred to this as “frequency of use analysis”.
175. Dr Camp’s written evidence was that a medicinal chemist would have undertaken this sort of work and would have been very interested in the results.
176. BMS’s position is best articulated in a series of steps. I have based the following on its closing written submissions.
177. First, it points to Embodiment 7 as being the first in `652 to define compounds necessarily incorporating a lactam, with a bicyclic core as follows:
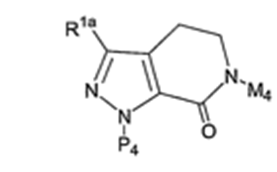
178. Here the lactam of interest is, BMS said, at M4 (there is also one in the core itself).
179. BMS then submits that in Embodiment 7:
179.1 M4 is defined as A-B which is selected from the following two substituents comprising a phenyl group attached to a lactam (`652 at page 68 lines 3-9):
179.2 P4 is defined as G (page 68 at line 1) which is itself defined as the group of compounds listed from line 6 of page 57; and
179.3 R1a is defined at page 52 lines 11-12.
180. It is worth mentioning that when it comes to the structural analysis below BMS says that M4 would be understood to bind in the S4 pocket of factor Xa and P4 in the S1 pocket, with R1a as part of the scaffold.
181. Dr Camp called the lactam on the left side of the two options above “Lactam 1” and that on the right “Lactam 7” in an analysis which he then did and which was set out in his exhibit NPC26. NPC26 covers 131 compounds, of which 74 were synthesised (those in Embodiment 8). Rather confusingly, Dr Camp referred to M4 as R3, to P4 as R2 and to R1a as R1 in his exhibit NPC26.
182. Once organised in this way it is possible to analyse, Dr Camp said, the pattern of what the patentee did. BMS submitted that of the 74:
182.1 lactam 1 was by far the most common lactam in the M4 position (42 instances, the next most common, lactam 7, having been used 24 times);
182.2 4-methoxyphenyl was by far the most common substituent in the P4 position (44 instances, the next most common, 3-chlorophenyl, having been used 6 times); and
182.3 CF3 was the most common substituent in the R1a position (20 instances, the next most common, carboxamide, having been used 13 times).
183. And it further submitted that the skilled medicinal chemist would realise that apixaban:
183.1 has the most common lactam in the M4 position, i.e. lactam 1;
183.2 has the most common substituent in the P4 position, i.e. 4-methoxyphenyl; and
183.3 has the second most common substituent in the R1a position, i.e. carboxamide.
184. This was very elaborate work, and one of its steps involved drawing the compounds starting from their names, so as to be able to identify what core and functional groups they had. The Claimants questioned whether CGK means existed at the priority date to do that, and whether the skilled medicinal chemist would undertake the exercise.
185. The issue of whether the tools existed to draw the structures was an unnecessary digression in my view and in any event I find that at least one software package existed that could do it (ChemDraw 6.0 Ultra), which the skilled medicinal chemist could find if they wanted to do the task.
186. Whether they would want to do the task to the extreme level of detail undertaken by Dr Camp is doubtful, in my view, but one has to bear in mind that Dr Camp was doing it in the crucible of litigation, where he had to take a rigorous approach given his obligations as an independent expert and because anything he said would be picked over most assiduously by the Claimants.
187. I accept Dr Camp’s evidence that the general scheme of what was done by the patentee and the compounds chosen for synthesis in terms of the patterns of structures could be identified with significantly less effort than went into NPC26. I also accept his evidence that with that lesser effort it would be appreciated, if apixaban were considered, that it was fairly typical, in the sense of using a core and substituents common to quite a large number of the compounds that were synthesised.
188. However, the utility of the analysis is quite another matter. I do not think there was any evidence that it was CGK to use this kind of frequency analysis to work out which compounds from a broad range were active, or promising. A crucial point to appreciate is that the analysis was done without any biological data to ground it. Dr Redshaw said, and I accept, that she had never analysed a set of compounds like this for which there were no biological data, and that there were many reasons why particular substituents might be frequently used, not just activity.
189. Dr Camp accepted in essence that lacking biological data the skilled medicinal chemist would not have done this sort of exercise and that the exercise was “just really understanding the issues that they [the authors of `652] are trying to resolve”. So he retreated a long way from his written evidence.
190. Even taking this analysis along with the indication from page 170 of `652 that some positive results existed does not help BMS. One simply cannot infer which if any of the 74 compounds had good biological results, which had bad results, and which had no results. Nor can one infer whether the “typical” compounds all behaved the same, or similarly. As Counsel for the Claimants put to Dr Camp at one point, this is SAR (structure-activity-relationship) analysis without any “A”.
191. The Sheehan paper to which I have referred above in relation to the 3g point was again put to Dr Camp on this part of the case. He accepted that it showed 25 compounds with a particular structural feature having been made, with consistently unpromising results. This supported the Claimants’ position for similar reasons. Again, and as with the 3g point, Dr Camp did not identify any of the review papers from the factor Xa inhibitor art deploying frequency of use to identify promising compounds.
192. I also thought that Dr Camp’s oral evidence illustrated that this part of BMS’s case was artificial in working backwards from apixaban specifically, and the later knowledge that it is indeed a potent factor Xa inhibitor. This was apparent from his explanation of his earlier involvement in the Canadian case, and the analysis in relation to R1a, where apixaban has a carboxamide, which is not the most common substituent.
193. This is the most complex part of BMS’s case and understanding it and explaining it is not assisted by the varying and different ways in which the parties developed and organised their cases.
194. As it seemed to me, BMS’s position had the following key elements:
195. First that the skilled team would have known from the CGK of the crystal structure of factor Xa that binding in the S1 and S4 pockets was very important. I have accepted that when dealing with the CGK.
196. Second, that DPC-423 was a very well-known compound in the CGK with good activity. That was also common ground.
197. Third, that based on a comparison with DPC-423 and/or in the light of the common general knowledge, the structure and individual groups in apixaban made it plausible that it would be effective in binding to and inhibiting factor Xa. Both sides referred to the following comparison of DPC-423 and apixaban:
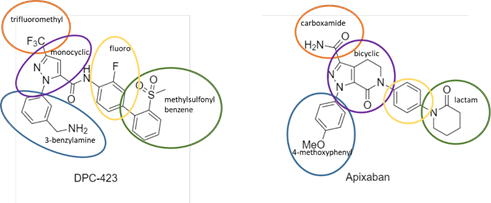
198. The Claimants submitted that DPC-423 was so different that it would not be called to mind at all when considering `652, but this comparison at least provides a way to organise the topics which need considering in relation to the structural argument. The topics of relevance are:
198.1 Apixaban has a 4-methoxyphenyl in the P1 position where DPC-423 has a 3-benzylamine.
198.2 Apixaban has a lactam at the P4 position where DPC-423 has a methylsulfonyl benzene.
198.3 Apixaban has a bicyclic core where DPC-423 has a monocyclic core. BMS called this apixaban’s “rigidified” core.
198.4 Apixaban has a carboxamide attached to the pyrazole ring (circled in orange) where DPC-423 has a trifluoromethyl.
199. I will deal with the points in that order. It was common ground that the additional fluoro group ringed in yellow was unimportant.
200. In his oral evidence, Dr Camp outlined a still more complex analysis which he called the “direction of travel” and which embraced `131 and other matters. BMS did not defend this.
201. There are two main strands to this point. The first is whether there was a basis in the CGK for this substituent as a S1 binder. The second is whether the chemistry of it would provide a basis for thinking it would be beneficial, or not.
202. The CGK basis relied on by BMS was the Lilly series identified above. I have held that they were CGK and that so was a more general move towards neutral binders at the P1 position. However, the fact that the series was CGK does not mean that it was well understood. Dr Camp’s written evidence said that its binding mode was unclear, and in his oral evidence he agreed that one of the review papers (Rai) did not regard the 4-methoxyphenyl as being the P1 element/S1 binder. Other of the review papers (Ries, Betz) proposed replacing the 4-methoxyphenyl group, and Maignan & Mikol expressed the same view.
203. Confronted with these papers, Dr Camp’s evidence was ultimately to the following effect (T4/319):
Q. Having looked at this Lilly series, and what it is said about
it, there was no oral bioavailability data as you agree, I suggest in the light of that, this Lilly series would not have been regarded by the medicinal chemist in 2001 as being a key series of inhibitors?
A. I think it is more just the groups, you know, the neutral S1
binder. I mean, clearly the Lilly series here is not very well optimised. I can just tell that looking at the structures. So I think it is more the fact it has a neutral S1 binder.
Q. In fact, in this sheer Ries [sic, series] we do not know what binds in S1 and what binds in S4, do we?
A. Not only based on modelling predictions, obviously it is a
weakly active compound and it has the same group, so I agree
it is not clear.
Q. No. If you had read what these papers have said about this
series, the medicinal chemist would not have had in mind the
para-methoxyphenyl group as being a successful binder, would
he?
A. Not based on this series.
204. So I reject the P1 4-methoxyphenyl case based on the Lilly series. Not only was the series not well optimised, but it was not even clear which way round it bound (it was agreed to be CGK that this kind of S1/S4 “flipping” was possible). Also, when being asked about the DPC-423 comparison more generally Dr Camp said that the Lilly series was “totally different” and not comparable, and I found this very hard to square with his relying on it for the 4-methoxyphenyl group.
205. As I have mentioned above, the DuPont series also relied on in this connection by Dr Camp in his written evidence was not CGK, and there were no data for it; BMS did not rely on it.
206. Further, when he was asked about a direct comparison between DPC-423 and apixaban at the P1 position, Dr Camp accepted that while the benzylamine at the 3 position in DPC-423 could form a salt bridge with Asp189 at the base of the S1 pocket, the 4-methoxyphenyl in apixaban could not. In this context he said that it was “very, very hard” to make a prediction, that it was not obvious why the 4-methoxyphenyl group would go in the S1 pocket and that from a structure-based design perspective it was “very unusual” that (as we now know) it did. He said “I do not think you can really rationalise it” and it was an unexpected finding. See T5/505-507.
207. Dr Camp had summarised his views on this point at paragraph 4.1 of his second report. One matter on which he and Dr Redshaw agreed and which he mentioned there was that the S4 pocket allowed for a degree of variability (was “catholic” as Dr Redshaw put it in oral evidence). While that helps BMS to some extent it does not mean that any group would be regarded as a plausible binder at that location.
208. Other of Dr Camp’s points in paragraph 4.1 were significantly undermined in cross-examination. In particular it turned out there was no CGK basis for 4.1(c) and the examples he had given did not support 4.1(d). When he was asked in general terms about the lactam (at T5/518) there was the following exchange:
Q. What I would suggest to you, doctor, having looked at your
reasons for saying that the medicinal chemist would expect the
lactam to bind in the S4 pocket, I suggest that there is
absolutely nothing here to support that conclusion and the
medicinal chemist would not, based on his common general
knowledge, be able to make a reasonable prediction that the
lactam group would bind in that S4 pocket?
A. I think the lactam has to go in that pocket. It cannot go
anywhere else. It is hard to rationalise it, I totally agree,
but looking at the binding modes, you know, it cannot go
anywhere else. So it has to go in the S4 pocket, and
obviously you can test that. You know, this is sort of, in my
opinion, well beyond the structure-based design. I think your
comments are fair.
209. What this reflected was that based on the later knowledge that apixaban does bind, one now knows that it must be the case that the lactam group binds in the S4 pocket. But even now it is hard to rationalise and it is well beyond structure-based design. Any possibility that `652 could provide plausibility based on structure is clearly excluded in respect of this group.
210. In comparison with the two previous points this one was relatively neutral; rigidification of this kind was generally known to be possible and might or might not be beneficial. It would introduce an extra degree of uncertainty in trying to make a prediction from DPC-423, however. There was no positive reason to think it would work and no positive reason to think it would not.
211. Dr Redshaw’s written evidence was that:
211.1 The compounds with 4-methoxyphenyl groups in the review papers would not have been of interest.
211.2 No prediction from the 4-methoxyphenyl compounds could be made, in particular because of the uncertainty of the orientation of binding to which I have referred above.
211.3 Although there was flexibility in the S4 pocket there was no reason to think that the lactam group would bind there.
211.4 There was no reason to think the rigidified core would hold a 4-methoxyphenyl and a lactam group in the right orientation.
212. Counsel for BMS made some progress on the first point and I have concluded that the Lilly compounds were CGK, but Dr Redshaw was not effectively challenged on the other three points and I accept Dr Redshaw’s evidence.
213. Taking all three differences from DPC-423, Dr Camp accepted that it was not possible to predict the properties of the synthesised compounds, even the ones with the 4-methoxyphenyl group (see T5/511).
214. I find that the overall position is that in relation to the individual points and in relation to their aggregate effect no prediction based on structure could be made from the CGK, and indeed apixaban’s binding is unexpected and hard to explain. There is nothing in the CGK positively to say that it could not bind effectively, but that is not the point - there has to be some positive reason to think there might be success.
215. It is also a significant problem for BMS that its case based on structure relies entirely on CGK. It does not draw on anything in `652 at all. So if plausibility were to be based on structure I cannot see how it represents a contribution by the patentee.
216. Taking all the above matters together, I conclude that `652 does not make it plausible that apixaban would have factor Xa binding of the level of 10 µM as referred to on page 170, or any useful degree of binding. The fundamental problem is that identified by the Claimants: there is simply no reference to apixaban there to allow an inference that it was one of the compounds for which useful results had been achieved. The frequency of use analysis suffers from the problems identified above and while the reader of `652 would infer that work of some kind had been done on lactams with quite a number made, there is no way to connect any particular compound to any degree of activity. Apixaban had been made in quantity but that does not mean anything for activity, and the structural arguments fail on the facts.
217. So BMS’s points fail individually and their whole is no greater than the sum of their parts. Since there is no plausibility of any meaningful factor Xa binding the Patent is invalid, since all the applications for apixaban depend on factor Xa binding. I will however go on to make conditional factual findings about those applications.
218. Even if `652 had made it plausible that apixaban had the degree of binding indicated on page 170 (10 µM), on my findings as to the CGK that would not make it plausible that it would be useful in therapy, because nanomolar potencies were needed for that.
219. `652 contains nothing to indicate that apixaban is selective for factor Xa as compared with other serine proteases. As I have indicated above, the Claimants said that this gave rise to an additional lack of plausibility because selectivity is needed in view of the fact that inhibition of other serine proteases may interfere with what would otherwise be a useful effect on factor Xa. The Claimants argued that this is not just about side effects (they accept that a patent does not have to exclude side effects to make therapy plausible) but about efficacy, for the treatment of the thromboembolic conditions in question.
220. However, against that `652 does not promise any such selectivity.
221. In my view, had it been the case that `652 made plausible a level of factor Xa inhibition which could form the basis of an effective therapy, an omission to prove selectivity would not mean plausibility for therapy could not be shown. My reason is that not showing selectivity would only mean that there was a risk of reduced overall efficacy by an off-target effect on another serine protease. It would not mean overall efficacy was not plausible. The statement in `652 that some activity against other serine proteases might be possessed by “compounds of the present invention”, to which I have referred above, does not change the fact that an off-target effect would be merely a possibility.
222. I accept BMS’s contentions that it would not have been difficult or burdensome to test apixaban for its factor Xa inhibitory activity, and that if such tests were done a very good level of activity would have been found. The same applies to selectivity and to bioavailability, although I have found above that lack of selectivity data would not lead to a lack of plausibility for therapy, and bioavailability would not be seen as essential since a drug could be given parenterally if necessary.
223. However, the fact that I accept BMS’s factual contentions about testing does not help it. In the absence of making some showing of plausibility based on one of the other matters relied on (the teaching on page 170, the 3g point, frequency of use, structure), the ability to test cannot get BMS any further than the patentee in Warner-Lambert. It provides (at a maximum) the sort of encouragement-plus-ability-to-test that the Supreme Court rejected, as I set out above. I say “at a maximum” because my analysis above means there is not even any encouragement concretely referable to apixaban.
224. The following non-therapeutic uses were relied on:
224.1 Use as standard or reference compounds.
224.2 Use in diagnostic assays.
224.3 Use as diagnostic agents and adjuncts, in particular as anti-coagulants where blood was to be kept in the fluid state for analysis or biological testing.
224.4 Use as “lead” compounds.
225. On the first day of trial a potential dispute arose about whether BMS had adequately pleaded reliance on non-therapeutic applications; after due consideration the Claimants decided not to take a pleading objection. They made clear that they disputed any non-therapeutic utility and they argued, as they surely were entitled to, that whatever the pleading position BMS had led no written evidence in support. In my view they were right about that and I note that BMS’s written closing submissions on this topic contained no meaningful references to its experts’ reports, with the exception of a single reference to Dr Camp’s report on lead compounds. It is possible that BMS’s pleading covered non-therapeutic uses (although I am very doubtful whether it did), but it certainly did not flag them up, and the fact that BMS’s evidence did not deal with it means that I think the Claimants were taken by surprise by it. The Claimants’ decision not to take a pleading point was, I expect, a pragmatic one because they wanted to avoid disruption to the trial and because they thought they could address non-therapeutic uses adequately.
226. There was also only limited cross-examination on non-therapeutic uses, to which I return below.
227. I have dealt with this above when dealing with the law. I do not think it can be a meaningful technical contribution for a compound to have such a low level of activity that it can serve as an example of, in effect, what not to do.
228. There was no evidence at all as to this from BMS’s experts in their written evidence. Prof Morrissey was asked about it in cross-examination and it transpired that he had minimal experience, if any, to speak from (to be fair, he had not claimed any). In any event he accepted that for the assay format he had in mind as a potential diagnostic use the purpose of using the factor Xa inhibitor would be to demonstrate that cleavage of a chromogenic or other substrate was being done by factor Xa and not by another enzyme such as thrombin. He accepted that this would require selectivity and of course `652 contains no pointer at all that any of the relevant compounds are selective. So unlike therapeutic use I conclude that lack of any showing of selectivity means a lack of plausibility for this potential application. In any event I found it very speculative.
229. The diagnostic assays point was not even put to Dr Leadley. The explanation offered was that Dr Leadley had not given any evidence on it. I reject that explanation - the reason Dr Leadley had not given evidence on it was that the Claimants were taken by surprise and it is perfectly possible that Dr Leadley would have had relevant views. The point should have been put if it was to be run.
230. There was some basis in the literature for this as a potential use for factor Xa inhibitors. Dr Leadley had referred to it in a review article in 2001 and (less convincingly) it was mentioned in an RPR patent application. BMS relied on the fact that the latter said that “any inhibitor of Factor Xa activity” would be suitable for the purpose, but I find that is clearly an incorrect overstatement.
231. Dr Leadley explained in cross-examination that anti-coagulant use would require a demonstration of prevention of clotting and that there was no reason to suppose that such use would require any lower activity than use in therapy; there was good reason to suppose that in e.g. glass vessels a higher degree of activity would be needed. I accept Dr Leadley’s evidence and my finding is fortified by the fact that Prof Morrissey largely agreed. He agreed that for this sort of application the skilled person would need to be reasonably convinced of factor Xa inhibition in the prothrombinase complex, in particular by reference to a PT or aPTT assay (not done in `652), and in that context he accepted that the idea of using compounds from `652 in this way was “speculation”. He did offer the idea that the need for a higher level of activity could be met by dosing “at a very high level” but I thought that too was very speculative.
232. As I have said above, “lead” compound means a starting point for research, not a compound that was already “leading” in the sense of ready to be used for a practical purpose. It is true that there was evidence that compounds with Kis of about 10 µM could be lead compounds in this sense, but nonetheless I think this is just a less extreme version of the reference compound argument. It is not a technical contribution for something to be lacking practical utility but to be a starting point for research that it might be hoped would lead to something which did have such utility.
233. BMS’s case in relation to non-therapeutic uses would fail on the facts for these various reasons even if (contrary to my earlier conclusion) the specification of `652 made it plausible that apixaban had some level of factor Xa inhibitory activity as indicated on page 170.
234. `131 was published on 6 July 2000. The Claimants pointed out that it was filed by DuPont Pharmaceuticals Co, whose business was later sold to BMS. This explains the commonality in teaching and approach of `131 and `652 (and there are also two inventors in common) but is not directly relevant to the legal issues I have to decide.
235. The title of `131 is “Nitrogen containing heterobicycles as factor Xa inhibitors”. I need not go through its teaching in as much detail as with `652. What matters for the purposes of the Claimants’ obviousness attack can be summarised quite shortly:
235.1 Apixaban is (it is common ground) embraced within the first, second, third, fourth, eighth and ninth embodiments of `131. Those embodiments are broadly defined and in none of them is apixaban individually identified.
235.2 An assertion very similar to that on page 170 of `652, of a Ki of 10 µM or less for some unidentified compounds, is made in `131 at page 264.
235.3 The compounds of `131 are taught to be useful as anticoagulants for treating thromboembolic disorders, at page 263.
235.4 Essentially the same teaching as to non-therapeutic applications as is in `652 is to be found in `131, at pages 264 and 267.
236. The Claimants’ attack based on `131 is one of lack of any technical contribution; I have already said that it is not a “classical” obviousness attack based on positive pointers or motivation to make apixaban or to think that it, of all the very many compounds falling within the teaching, would be likely to be a useful factor Xa inhibitor. I mention that simply because BMS repeatedly pointed out that a classical obviousness attack would have failed, a stance which it maintained in its written closing submissions; but that was just a distraction from the real attack. I record that BMS pointed out that EPO case law (see e.g. T 184/16) is that the standards for plausibility and obviousness are not the same. This can be useful for patentees fighting off a squeeze between classical obviousness and insufficiency but is not relevant to the attack based on `131.
237. In identifying the relevant disclosure of `131 I have pointed out that it contains the same teaching as `652 in relation to utility, whether in terms of factor Xa inhibitory activity levels, therapy, or non-therapeutic uses. However, that teaching is referable to different classes of compounds than in `652, and while the classes cover apixaban, they do not disclose it.
238. I have found that `652 lacks plausibility within its own terms, without the need to consider any prior art. It is therefore bound to lack any technical contribution over `131 and in that very limited sense this attack succeeds.
239. However, if I am right in what I say above about plausibility then the Patent is invalid anyway. The attack over `131 would only matter if I were wrong about plausibility. Although the Claimants maintained that the attack over `131 is a distinct one, I found it hard to see a realistic scenario in which it would succeed if the plausibility attack failed. In the end, I think their point was that if `652 were to be plausible based purely on teaching that was in `131 (albeit in relation to different classes of compounds) then the plausibility attack would fail but the attack over `131 would succeed because there could be no invention in just picking different compounds from within `131 (including apixaban) without their being any better.
240. However, I cannot see that there is any realistic way in which plausibility of `652 could succeed based purely on teaching that is in `131. I do not think that was even argued: BMS’s case depends on the patentee in `652 having named apixaban as a specific compound which had been made in quantity, such that it could be inferred that the patentee thought it was promising, bolstered by its being a “typical” lactam, and the structural case. That information is not in `131 (nor is the structural case in `652, of course - it is from the CGK as I explained above).
241. So while this attack succeeds in the limited sense identified above, it does not add anything and it does not require any additional factual findings. If this matter were to go on appeal I do not suppose the attack over `131 will be considered if I am right about plausibility. If I am wrong, the appeal court will be able to assess the attack over `131 with my factual findings and, crucially, an appreciation of why I am wrong.
242. Teva makes a further obviousness case based on lack of technical contribution. In a nutshell, its argument is that given that the structure of apixaban was obvious, in that (as I understood the argument) there was no invention necessary to make it, the only possible technical contribution could lie in its use for a particular purpose. So Teva says that any claim not limited to a particular use made plausible by the specification would be invalid for exceeding the technical contribution.
243. Teva said in closing written submissions that that scenario could arise and be important in the present case if BMS were to win in relation to plausibility on non-therapeutic uses but lose on plausibility for therapy. In its opening written submissions Teva had focused on the reverse case - that if apixaban was plausible for therapy then BMS should be limited to a claim limited to use in therapy and claims 1 to 6 were therefore invalid. So at that stage the argument could not have been decisive to the result of the action as it would not have knocked out claim 7.
244. Sandoz did not support this argument.
245. BMS responded that the argument must be wrong because it would mean that there could never be a claim to a novel compound per se, which it said was contrary to e.g. Generics v. Lundbeck [2009] RPC 13 at 71-72.
246. Teva did not contend that any UK authority supports the argument directly, although it said that Warner-Lambert and Biogen v. Medeva [1997] RPC 1 were thematically consistent with it and that there was no authority directly contrary to it. Teva also did not say that there was EPO authority to support the argument.
247. This is a radical argument which could have far-reaching effects. At the start of the case it did not seem likely to be of practical importance. It was only lightly argued to me. Since it does not matter to the result because I have found the Patent invalid for lack of any plausibility, and since it involves no further factual questions, I decline to decide it. I am particularly concerned at the prospect of deciding it without a clear view of whether it is consistent with EPO case law and indeed other decisions under the EPC in other jurisdictions.
248. As I have mentioned in the introduction to this judgment, BMS proposes amended claims 7A and 7B which add the requirements that the claimed compound be “a factor Xa inhibitor” (proposed amended claim 7A) or “an effective factor Xa inhibitor” (proposed amended claim 7B).
249. The purpose of these amendments, as I have already said, was to bring the claims more directly in line with those considered in Fibrogen should that be necessary. BMS said that if it were necessary, the amendments would result in a claim in which factor Xa inhibition was a step one functional feature and treatment of a thromboembolic disorder was a step two functional feature. In the light of my findings above that there is no plausibility for factor Xa inhibition or usefulness in therapy, the amendments would not make any difference and they would not cure the invalidity I have found, but I will decide their formal allowability.
250. The UKIPO considered the proposed amendments and raised no objection.
251. The Claimants opposed the amendments on the basis that there was added matter because the application did not disclose apixaban specifically as a factor Xa inhibitor, and on the basis that there was a lack of clarity. The lack of clarity was said to be that neither proposed claim 7A nor proposed claim 7B was clear as to the level of inhibition needed. I was also concerned (and the Claimants argued in closing) that if claim 7A and claim 7B were both allowed then there might be confusion about what “effective” added but this fell away when Counsel for BMS explained that the two forms were sought in the alternative.
252. In relation to added matter, I consider that there is basis for the proposed amendments from, in particular (using references to `652), page 7 lines 2 to 5 which refers to lactams as factor Xa inhibitors, from Embodiment 8, and from claim 8 which is to each of the individual compounds of Embodiment 8 including apixaban. The application as a whole has a consistent emphasis on therapy, with the therapeutic effect taught to arise from factor Xa inhibition. I make it clear that these were put forward as disclosures of factor Xa inhibition in the sense only of asserting it, not as information rendering it plausible; Counsel for BMS did not argue that if there was a disclosure adequate to avoid added matter, then that also necessarily passed the test for plausibility.
253. The Claimants did not really press any added matter objection at trial.
254. As to clarity, I do not think that the lack of a numerical limit defining the necessary level of inhibition means that there is a problem. Many claim features are qualitative rather than quantitative and although there could be a debate how much inhibition was necessary for e.g. therapeutic effect in a given case that does not mean the claim is insufficiently clear.
255. In my view, of the two options put forward by BMS proposed amended claim 7A is preferable as corresponding more closely to the passage providing basis on page 7.
256. Therefore, I find that proposed amended claim 7A would be formally allowable but I refuse the amendment on the basis that it would not cure the invalidity that I have found.
257. I conclude that:
257.1 The Patent, European Patent (UK) 1 427 415 B1, is invalid by reason of lack of plausibility.
257.2 The attack of obviousness over `131 for lack of technical contribution also succeeds but does not add anything.
257.3 I decline to decide Teva’s obviousness attack based on the claims exceeding the technical contribution.
257.4 The proposed amendments to the Patent are formally allowable but do not cure the invalidity.
257.5 Because the Patent is invalid, so is SPC/GB11/042.
258. I will hear Counsel as to the form of Order if it cannot be agreed. I direct that time for seeking permission to appeal shall not run until after the hearing on the form of Order (or the making of such Order if it is agreed).
ANNEX A to Judgment of Meade J of 7 April 2022 in Actions HP-2020-000042 and HP-2021-000003
[2022] EWHC 822 (Pat)
Sandoz and Teva v BMS
AGREED CGK
Claim Nos HP-2020-000042 and
HP-2021-000003
IN THE HIGH COURT OF JUSTICE
BUSINESS AND PROPERTY COURTS OF ENGLAND & WALES
INTELLECTUAL PROPERTY LIST (ChD)
PATENTS COURT
B E T W E E N:
SANDOZ LIMITED
Claimant/Part 20 Defendant in HP-2020-000042
and
BRISTOL-MYERS SQUIBB HOLDINGS IRELAND UNLIMITED COMPANY
(a company incorporated under the laws of Ireland)
Defendant/Part 20 Claimant in HP-2020-000042
(the “Sandoz Action”):
TEVA PHARMACEUTICAL INDUSTRIES LIMITED
Claimant in HP-2021-000003
and
BRISTOL-MYERS SQUIBB HOLDINGS IRELAND UNLIMITED COMPANY
Defendant/Part 20 Claimant in HP-2021-000003
and
TEVA UK LIMITED
Part 20 Defendant in HP-2021-000003
(the “Teva Action”):
_______________________________________
STATEMENT OF AGREED COMMON GENERAL KNOWLEDGE
_______________________________________
We submit the following facts and matters represent the common general knowledge (“CGK”). This document records the agreed common general knowledge of the skilled team. Identification of paragraphs in the expert evidence (in square brackets) is provided for reference only and is not intended to incorporate additional agreed facts.
Thrombosis
123. Thrombosis is the formation of an unwanted blood clot (termed a “thrombus”) inside a blood vessel. Thrombosis is one of the leading causes of disability and death in the world. An intravascular thrombus can grow to completely block a blood vessel, leading to ischemia and death of downstream tissues. If blood clots form in arteries and block the flow of blood, it can lead to a heart attack, stroke, or lower limb gangrene requiring amputation. Blood clots that form in the deep veins of the legs, known as deep-vein thrombosis (“DVT”), can break off, travel round the circulatory system (embolize) and lodge in an artery of the lungs, causing a pulmonary embolism (“PE”). A clot, or a piece of the clot, that breaks free and begins to travel around the body is known as an embolus. [Morrissey 1/6.6; Leadley 1/5.6]
124. In healthy humans, homeostatic balance exists between procoagulant (clotting) forces and anticoagulant and fibrinolytic forces. Under normal physiological conditions, intravascular blood should flow freely and not clot. It is only on injury that the coagulation system should respond rapidly and locally at the site of injury to a vessel wall to generate a clot. As noted above, unwanted intravascular clotting is termed thrombosis. Too little clotting, on the other hand, results in disorders of excessive bleeding termed hemorrhagic disorders such as hemophilia. [Leadley 1/5.8 & 5.28]
125. Numerous genetic and environmental factors, aging, certain medical conditions (such as cancer and autoimmune disease), and surgical procedures, can all tip the balance in favor of coagulation, leading to the pathologic formation of thrombi in blood vessels. [Leadley 1/5.29]
126. “Thrombotic disorder” usually refers to a range of disease states in which thrombi form inside blood vessels, which pose the threat of obstructing blood circulation. The term “thromboembolic disorder” can be used as a general term that encompasses both thrombi that form in place as well as thrombi that have broken loose and lodged in a blood vessel in another part of the circulatory system. [Morrissey 1/6.7; Leadley 1/5.29]
127. It was well known that thrombotic/thromboembolic disorders could be treated by reducing blood clotting through inhibiting the coagulation cascade. [Morrissey 1/6.9; Leadley 1/5.8] In 2001, the goal for antithrombotic drugs was preventing thrombosis while retaining hemostasis (explained further below) i.e., preventing intravascular clots without causing bleeding complications. [Leadley 1/5.7]
The coagulation cascade
128. In a healthy human being, clotting processes prevent excessive bleeding and participate in repair of damaged blood vessels. If the system is functioning properly, blood clots that form at the sites of blood vessel injury seal the leaks and are then removed later on. Hemostasis is the mechanism that leads to cessation of bleeding from a blood vessel. This is critical for survival by preventing uncontrolled blood loss (or hemorrhage) which would result from even minor injuries in the absence of clotting. [Morrissey 1/6.8; Leadley 1/5.5]
129. Humans have evolved a complex regulatory system which ensures unimpeded blood flow under normal, physiological, conditions, but with the ability to rapidly respond to damage to the blood vessels through a process that involves multiple interlinked steps culminating in the formation of a “hemostatic plug” that closes up the damaged site of the blood vessel, controlling the bleeding. There are two stages of hemostasis:
(a) Primary hemostasis refers to platelet aggregation and platelet plug formation. Platelets circulate in the blood in inactive form. The platelets become activated, for example, by coming into contact with collagen on the vessel wall (exposed by damage), or by the action of thrombin. Activation causes platelets to clump together and adhere to the site of injury and to each other, plugging the injury.
(b) Secondary hemostasis refers to the process in which fibrin is formed to stabilize the loose platelet clot formed in primary hemostasis. Secondary hemostasis involves a cascade of enzymatic reactions (the “coagulation cascade”) that ultimately results in the conversion of fibrinogen to fibrin monomers. Fibrin monomers then self-associate into fibrin strands which are then cross-linked into insoluble strands that serve to stabilize the loose platelet clot formed in primary hemostasis. [Morrissey 1/6.8; Leadley 1/5.5]
130. The coagulation process involves a complex set of reactions involving approximately 30 different proteins . The enzymes involved in the coagulation cascade are termed “factors” and are referred to by Roman numerals as factors I-XIII. The “a” in the name of a factor indicates the factor in its active form. The coagulation cascade can be considered as a series of activation steps, each of which involves a proteolytic conversion of a zymogen (the inactive precursor of an enzyme, in this case the inactive form of the clotting factor, many of which are produced by the liver and secreted into the circulation) in response to injury or tissue damage, to the corresponding active serine protease (explained further below). The active form of each clotting factor converts the inactive form of the next factor in the coagulation cascade to its active form (although some factors have multiple roles). [Morrissey 1/6.10; Morrissey 2/2.2; Leadley 1/5.8 & 5.9]
131. Almost all of the active enzymes in the coagulation cascade are serine proteases and are structurally related to the digestive enzyme trypsin. Protease is the term for an enzyme which catalyses the cleavage of a peptide bond. Serine proteases are so-called because they have the amino acid serine in their active site. Serine proteases cleave peptide bonds by using a hydrolysis reaction to break down large proteins into smaller peptides. For coagulation factors, this cleavage results in the exposure of the active site of the enzyme, thereby activating the zymogen. Serine proteases are ubiquitous and involved in multiple physiological processes including digestion, development, fertilization, apoptosis and immunity, in addition to coagulation and the related process of fibrinolysis (described below). [Leadley 1/5.10]
132. There are two main pathways for triggering the coagulation cascade: the intrinsic (or contact) pathway and the extrinsic (or tissue factor) pathway. These merge to form a third pathway: the common pathway. [Morrissey 1/6.11; Leadley 1/5.8]
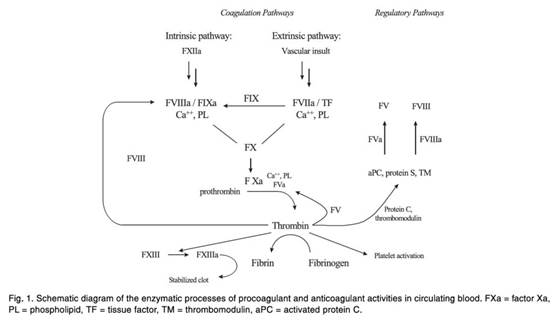
Figure 1 Blood coagulation cascade and sites for antithrombotic therapy [Leadley 1/Figure 1]
133. The intrinsic pathway is so-called because all the necessary components of this pathway are in the plasma and no external source is required to trigger this pathway (unlike the extrinsic pathway that, as explained below, requires exposure to tissue factor for triggering). The intrinsic pathway only plays a limited role in hemostasis, but is initiated by the activation of factor XII when blood comes into contact with certain negatively charged surfaces, such as glass, or is exposed to endothelial collagen, which occurs when tissue damage occurs. The intrinsic pathway ultimately results in the generation of factor XIa, which then converts factor IX to fIXa which activates factor X to factor Xa. [Morrissey 1/6.12; Leadley 1/5.11]
134. The extrinsic pathway is activated when the blood comes into contact with tissue factor (also called factor III). The complex of tissue factor and factor VIIa initiates the coagulation cascade by activating either factor IX or factor X. [Morrissey 1/6.14; Leadley 1/5.13]
Factor Xa
135. As explained above, the intrinsic and extrinsic pathways converge in the common pathway with the activation of factor X to factor Xa. Once activated, factor Xa goes on to activate factor II (prothrombin) to factor IIa (thrombin). While it is possible for factor Xa to do this alone, the rate of reaction is very low. Instead, the majority of prothrombin conversion into thrombin is catalysed by the formation of the prothrombinase complex (factor Xa, factor Va and calcium ions and an activated platelet surface). [Morrissey 1/6.15; Leadley 1/5.14]
Thrombin
136. Thrombin is a serine protease that plays a central role in thrombosis and hemostasis. It is the terminal protease in the coagulation cascade, and it is directly responsible for cleaving soluble fibrinogen to insoluble fibrin. Fibrin subunits then come together to form fibrin strands, and factor XIII acts on fibrin strands to form a fibrin mesh. This mesh helps to stabilize the platelet plug which forms the clot and also contributes to their attachment to the vessel wall. [Morrissey 1/6.16; Leadley 1/5.15]
137. Thrombin is also a potent stimulator of platelet activation. Activated platelets bind to fibrinogen and form cross-bridges to other platelets leading to platelet aggregation and thrombus growth. [Leadley 1/5.16]
138. The platelet and coagulation mechanisms are linked because the enzymatic reactions of the coagulation cascade predominantly take place on the surface of activated platelets. [Leadley 1/5.17]
Other co-factors
139. Other co-factors involved in coagulation include tissue factor, factor V, factor VIII, and high-molecular-weight kininogen. Calcium ions are also required to form the tenase and prothrombinase complexes which localize coagulation factors to the surface of activated platelets and act to accelerate the process of coagulation. [Leadley 1/5.19]
Physiological regulators of coagulation
140. The process of thrombin generation must be localized and contained to prevent widespread clot formation in the vascular system. Therefore, to control the process of coagulation, activated enzymes have naturally occurring inhibitors. [Leadley 1/5.20]
141. Proteins C and S act to prevent coagulation. Protein C is activated proteolytically by the thrombin/thrombomodulin complex, converting it to aPC. aPC inhibits coagulation by inactivating factors Va and VIIIa, with protein S acting as a co-factor. [Leadley 1/5.23; Leadley 3/1.9]
142. Tissue Factor Pathway Inhibitor (TFPI) is an anticoagulant protein that inhibits the extrinsic pathway by inhibiting factor VIIa in a mechanism that requires factor X and calcium ions and also directly inhibits factor Xa. [Leadley 1/5.24]
Fibrinolysis
143. Fibrinolysis is a normal process that prevents intravascular thrombi from growing and is essential for re-establishing normal blood flow. The process is mediated by plasmin, which is present in the circulation as an inactive precursor, plasminogen. Like the factors of the coagulation cascade, plasminogen is a zymogen which is converted to the serine protease, plasmin, once activated. Plasminogen activation takes place either by the action of tissue plasminogen activator (tPA) or urokinase. [Leadley 1/5.25]
144. At initial stages of thrombus formation during hemostasis, thrombus growth is necessary so plasminogen activators, such as tPA, are inhibited. However, as the structural integrity of the blood vessel wall is restored, endothelial cells begin to secrete tPA to slow the growth and initiate the breakdown of the thrombus. [Leadley 1/5.26]
145. Fibrinolytic drugs that convert plasminogen to plasmin are used to treat acute, life-threatening thrombotic disorders, such as heart attack and ischemic stroke. [Leadley 1/5.27]
Anticoagulants
146. Anticoagulants are a class of antithrombotic drugs which inhibit the formation and propagation of intravascular thrombi and are used in the treatment and prevention of many conditions. They are commonly used in patients who have experienced a first thromboembolic event or who are predisposed to developing a thromboembolic disorder, e.g., because they are undergoing surgery or have been diagnosed with atrial fibrillation which increases the risk of stroke. Anticoagulants may be administered parenterally via intravenous or subcutaneous injection, or orally. For reasons of patient convenience, safety, and compliance, oral anticoagulants are preferred, and were in 2001, for long-term out-patient management of chronic conditions. [Leadley 1/5.30]
147. In 2001, the main anticoagulants in use in the clinic were heparin (administered parenterally) and warfarin (administered orally). These are discussed further below. [Leadley 1/5.31]
148. Anticoagulants are usually prescribed when either the patient already has a blood clot (with the aim of stopping the clot increasing in size and embolising thus giving the body a chance to break down the clot itself over time) or the patient is at high risk of developing a blood clot (with the aim of preventing a blood clot forming in the first place). [Morrissey 1/6.19]
Vitamin K antagonists
149. Vitamin K antagonists (“VKAs”) block the processing of vitamin K in the liver. VKAs have been in use since the 1950s and one very well-known example is warfarin (discussed below). [Morrissey 1/6.20; Leadley 1/5.18]
150. A number of coagulation factors are synthesized in the liver using vitamin K, such as prothrombin and factors VII, IX and X. Therefore, by blocking vitamin K, the synthesis and proper post-translation modification of these vitamin K-dependent proteins is slowed down. Administration of VKAs results in the reduction in the levels of active prothrombin and factors VII, IX and X which play crucial roles in the coagulation cascade. VKAs also inhibit the proper post-translational modification of three anticoagulant proteins (proteins C, S and Z), affecting their Ca2+ and membrane binding capabilities (preventing enzyme complexes from forming). [Morrissey 1/6.21]
151. VKAs are often referred to as being indirect anticoagulants as they have no intrinsic anticoagulant activity themselves, but have an indirect effect on the coagulation cascade. In 2001, VKAs were the most widely used anticoagulants (and indeed were the only approved oral anticoagulants available at that time). [Morrissey 1/6.22]
Warfarin
152. Warfarin was first used commercially as rat poison, mediating its effects by preventing coagulation so that the rats died of internal bleeding. It was approved for medical use in the 1950s. [Leadley 1/5.39]
153. Warfarin (and VKAs in general) has several disadvantages:
(a) Slow onset of action (3-4 days). This is a problem if patients need rapid anticoagulation to treat e.g. pulmonary embolism, deep vein thrombosis or transient ischemic attack (also referred to as a mini stroke).
(b) Slow offset of action (3-4 days). This can be a problem if patients on warfarin suffer an injury or need emergency surgery.
(c) Food interactions, particularly in relation to vitamin K and alcohol, which result in variability in patient responses. The effect of warfarin can be reduced or abolished by vitamin K present in green vegetables, green tea, health foods, or nutritional supplements. Major changes in diet can significantly change a patient’s response to warfarin.
(d) Adverse drug-drug interactions which are particularly problematic in elderly patients who may need to take multiple medications.
(e) High patient-to-patient variability in dose which means patients need frequent monitoring to find and maintain the correct therapeutic window. Initially this may be every 1-2 weeks and thereafter at least every 12 weeks once a stable dose has been established for the patient. Older patients and those with co-morbidities may need routine monitoring every 1-2 weeks.
(f) Potentially serious side effects including hemorrhage (bleeding) and calcification of the patient’s arteries.
(g) Warfarin is teratogenic, meaning it harms a developing fetus and therefore is of particular concern if women treated with warfarin become pregnant.
[Morrissey 1/6.23, with respect to VKAs; Leadley 1/5.40]
154. VKA treatment was time consuming and costly, inconvenient for patients and still left patients with a significant risk of bleeding side effects. As a result, there was a desire to develop other anticoagulants. [Morrissey 1/6.24]
Heparins
155. Heparin, discovered in 1916, is a naturally occurring glycosaminoglycan which has been used as an anticoagulant since the 1930s [Leadley 1/5.32]. Heparins are used to treat pulmonary embolism, deep vein thrombosis, unstable angina, and heart attack, and are administered prophylactically to prevent thrombosis in a wide variety of medical and surgical situations. Heparin was the other anticoagulant that had been in widespread use for decades by 2001. [Morrissey 1/6.25; Leadley 1/5.33]
156. There are two main types of heparin drugs: (i) unfractionated heparin (“UFH”), also known as standard heparin; and (ii) low-molecular-weight-heparins (“LMWH”). Heparin must be administered subcutaneously or intravenously and so is more difficult to administer than VKAs. Heparin takes effect more quickly than VKAs, so it is usually given in clinical situations where an immediate effect is required. It is also possible to reverse the effects of UFH quickly using a reversal agent, protamine sulfate. [Morrissey 1/6.25; Leadley 1/5.34 & 5.35]
157. Heparin is naturally occurring and acts by binding to the enzyme inhibitor Antithrombin III (“AT” ). A deficiency in AT predisposes a person to thrombotic disorders. Heparin produces its major anticoagulant effect by inactivating thrombin and factor Xa through an AT-dependent mechanism. The catalytic-site serine of thrombin reacts with AT to form an inactive complex which prevents the thrombin from activating fibrin. Heparin is therefore an indirect inhibitor of thrombin and factor Xa. [Morrissey 1/6.26]
158. UFH is a sulfated polysaccharide with a molecular weight range of 3000 to 30 000 Da (mean, 15 000 Da). UFH suffers from the following drawbacks:
(a) it is poorly absorbed from the gastrointestinal tract and must therefore be administered by injection;
(b) it does not effectively inhibit prothrombinase activity;
(c) the size of the AT-heparin complex renders it incapable of inhibiting thrombin once it is in a complex with fibrin in a growing thrombus i.e., it has a very limited ability to access clot-bound clotting enzymes;
(d) it gives widely varying responses and a lack of predictability, requiring continuous monitoring of the patient;
(e) it has a fast onset of action and a short half-life (approximately 1 hour) and as a result, it must be given frequently or by a continuous infusion. This is possible in a hospital setting but less suitable for long-term use by patients at home;
(f) around 3% of those treated develop heparin-induced thrombocytopenia (“HIT”), which can be life-threatening;
(g) Side effects include hemorrhage (bleeding). Hemorrhage is a particular risk as, in addition to its action as an anti-coagulant, heparin also indirectly inhibits platelet function (via its ability to inactivate thrombin); and
(h) it can result in thrombotic rebound phenomenon after treatment is stopped.
[Morrissey 1/6.27; Leadley 1/5.34 & 5.38(c)]
159. In the 1980s, LMWHs were developed to try and overcome some of the problems associated with UFH and were found to be superior compared to UFH in several thrombotic indications and have the advantage of day-to-day dosing. LMWHs are fragments of UFH produced by controlled enzymatic or chemical depolymerization processes that yield chains with a mean molecular weight of about 5000 Da (i.e., approximately one-third the size of UFH). LMWHs were commercially available well before 2001. Unlike UFH which targets both thrombin and factor Xa, LMWH tends to favor factor Xa as a target over thrombin (owing to differences in the mechanism of action). [Morrissey 1/6.28; Leadley 1/5.35]
160. LMWH has several advantages over UFH: (i) it allows predictable and well-controlled anticoagulation, with fixed dose administration; (ii) its bioavailability and half-life is good with subcutaneous administration and does not require continuous monitoring; and (iii) patients are at reduced risk of developing HIT. However, the use of LMWH is not superior in every situation. Both types of heparin continue to be used in the clinic. [Morrissey 1/6.29; Leadley 1/5.38]
161. Another subset of heparins which had been developed by 2001 were the pentasaccharides. These were synthetic heparin fragments with even lower molecular weights than LMWH. By 2001, the best-known of these was “fondaparinux” (also referred to as Org31540 or SR90107A in the literature). Fondaparinux binds to AT in the plasma to have an anticoagulant effect and once bound to AT it only inhibits factor Xa and not thrombin. By 2001, fondaparinux was in phase III clinical trials and had reported positive results. [Leadley 1/5.50; Morrissey 1/6.30 - 6.32]
Hypocoagulation or bleeding risk
162. When patients are treated with anticoagulants, systemic hypocoagulation can occur where clots take too long to form. For this reason, anticoagulants are sometimes referred to as “blood thinners.” However, anticoagulants do not make the blood “thinner,” rather they prolong the time it takes for blood to form a clot. As a result, patients on anticoagulants can bleed easily and suffer spontaneous internal bleeding that can manifest as excessive bruising, gum bleeds, nose bleeds, and in serious cases, intracranial bleeding which can cause a stroke. These side effects are a direct result of the therapeutic action of anticoagulants and can outweigh the benefits of decreasing thrombotic risk. Weighing these risks is particularly important when considering preventative long-term treatment with anticoagulants. [Leadley 1/5.42]
163. Patients treated with heparin and warfarin therefore need regular monitoring to adjust their dose to ensure antithrombotic efficacy and prevent hypocoagulation. This monitoring involves measurements of how long it takes the blood to clot when activated in vitro; typical measurements employed are the prothrombin time (PT) and the activated partial thromboplastin time (aPTT) tests. [Leadley 1/5.43]
The need for a new generation of anticoagulants
164. It was well-established by 2001 that there was a clinical need to provide alternative oral anticoagulants to replace warfarin. [Leadley 1/5.44]
165. In particular, the clinical need in 2001 was for an oral antithrombotic that:
(a) Was effective at preventing thrombotic disease. It would be even more desirable if the drug was effective at preventing further growth of existing clots;
(b) Was safe and non-toxic;
(c) Had minimal side effects, in particular with respect to bleeding;
(d) Could be given orally once, or at most twice, a day. This would make it convenient for patients outside of hospital settings who were taking the drug long term, and therefore would be expected to affect patient compliance;
(e) Had low interpatient variability, including low drug-drug and low drug-food interactions;
(f) Had no need for frequent patient monitoring
[Leadley 1/5.45]
166. To achieve this, the drug would ideally have to be:
(a) A potent inhibitor of an appropriate target in the coagulation cascade;
(b) Selective (over other enzymes with important physiological functions both in the coagulation cascade and in other physiological processes);
(c) Orally bioavailable;
(d) Suitable for once or at most twice daily dosing; and
(e) Have a pharmacokinetic and pharmacodynamic profile which provides fast onset of action and short offset of action (hours rather than days, as with warfarin) in the event that patients require surgery, for example
[Leadley 1/5.46]
Serine protease inhibitors
167. Many of the coagulation factors are serine proteases (with exceptions including TF and factors V, VIII and XIII). Serine protease inhibitors interact with serine protease enzymes and reduce their activity by influencing the binding of substrate and/or the number of reactions the enzyme turns over per unit time. [Morrissey 1/6.33]
168. From the 1980s, many pharmaceutical companies focused resources on developing specific inhibitors of enzymes in the coagulation cascade. By 2001, there was very substantial interest in directly targeting the serine proteases in the coagulation cascade, in particular thrombin and factor Xa, and they were being actively pursued as targets for developing anticoagulants. The isolation from medicinal leeches of the naturally occurring direct thrombin inhibitor hirudin and the direct factor Xa inhibitor antistasin provided evidence that thrombin and factor Xa were viable drug targets. [Morrissey 1/6.34; Leadley 1/5.47]
169. Due to its proximity in the pathway to the blood clotting event, direct thrombin inhibitors were an early focus of drug discovery efforts to find new anticoagulant medicines. Thrombin had been heavily studied in the 1980s and early 1990s and its crystal structure was reported in 1989. Since thrombin plays a central role in the coagulation and platelet activation processes, by 2001 its regulation and activity had been studied in great detail in order to discover agents which would prevent thrombosis, ideally without substantially altering normal hemostasis. [Morrissey 1/6.35; Leadley 1/5.48]
170. However, thrombin’s complex role in blood coagulation, behaving as both a procoagulant and anticoagulant, as well as having an effect on platelet function, meant it was a difficult target to safely and effectively treat thrombosis [Leadley 1/5.48]. Direct thrombin inhibitors were thought to suffer from a number of potential drawbacks relative to factor Xa inhibitors:
(a) Inhibition of the prothrombinase complex (by targeting factor Xa) should prevent the continuing production of thrombin while maintaining a basal level of thrombin activity necessary for primary hemostasis. The prothrombinase complex present at the site of injury is unaffected by direct thrombin inhibitors and so cannot prevent the continuing production of thrombin;
(b) Thrombin inhibitors showed a tendency to increase the likelihood of bleeding complications; and
(c) Thrombin has both procoagulant actions (including converting fibrinogen into fibrin, activating factor XIII to XIIIa, and activating platelets) and anticoagulant actions (in the presence of thrombomodulin, converting protein C into activated protein C - an important natural anticoagulant in plasma). Given the known pleiotropic effects of thrombin, there was a concern that inhibiting thrombin’s enzymatic activity may have in vivo effects that would be difficult to predict.
[Morrissey 1/6.36]
171. Once thrombin has been formed, it can exert its coagulant effect by cleaving fibrinogen into fibrin before the inhibitor can bind to thrombin and exert its inhibitory effect. Factor Xa is the sole enzyme responsible for activation of prothrombin to thrombin and one molecule of factor Xa has been estimated to catalyze the formation of thousands of thrombin molecules. Therefore, a potential advantage of inhibiting factor Xa is that it may not need to be administered at as high a concentration as a thrombin inhibitor. Blocking one factor Xa molecule in effect prevents the formation of thousands of thrombin molecules. Lower doses would be expected to result in less systemic hypocoagulation, i.e., less bleeding. In addition, several studies have demonstrated that factor Xa inhibitors, compared to other mechanisms of inhibition of thrombus formation, produce antithrombotic effects at doses which only modestly alter markers of systemic hypocoagulation or bleeding. Consequently, many companies pursued the strategy to prevent thrombin formation by inhibiting factor Xa. [Leadley 1/5.49]
172. Factor Xa was identified as a promising target for the development of new synthetic anticoagulants following the isolation and characterisation in the late 1980s of the first naturally occurring specific factor Xa inhibitor, antistasin, isolated from leeches, and Tick Anticoagulant Peptide (TAP). TAP is a potent and specific inhibitor of factor Xa which inhibits thrombosis without causing excessive bleeding. [Leadley 1/5.50]
173. By 2001, essentially all the major pharmaceutical companies were attempting to discover novel factor Xa inhibitors including Merck, Corvus Pharmaceuticals, Du Pont, Bristol-Myers Squibb, Yamanouchi, Daiichi Sankyo, Hoechst Marion Roussel, LG Chem, Bayer, Johnson & Johnson, Schering AG, Rhône-Poulenc Rorer, Eli Lilly, Berlex and AstraZeneca. A variety of potent, selective, small molecule factor Xa inhibitors had been described in the scientific literature and some had been taken forward to clinical trials. [Leadley 1/5.51]
174. Factor Xa was considered to be a promising target with several potential advantages:
(a) Both the extrinsic and intrinsic pathways of coagulation culminate in factor Xa activation. Factor Xa then triggers thrombin generation and fibrin formation via the common pathway. Due to its position at the convergence of the two separate pathways and because it catalyzes the conversion of prothrombin to thrombin, factor Xa was understood to play a central and crucial role in the coagulation cascade;
(b) Factor Xa inhibitors were predicted to have a lower risk of bleeding than heparin and VKAs and a much wider therapeutic window than direct thrombin inhibitors because they specifically inhibit coagulation without directly affecting platelet function;
(c) Unlike thrombin, factor Xa was not thought to have functions outside the coagulation cascade and therefore negative side-effects as a consequence of inhibition were hoped to be limited; and
(d) When the clotting process begins, many molecules of factor X are activated and each factor Xa molecule can activate more than one substrate molecule. In fact, it was known in 2001 that one molecule of factor Xa could generate many molecules of thrombin per minute. It was therefore hypothesized that factor Xa inhibition could be a more effective and safer way to prevent blood clot formation than direct thrombin inhibitors as less drug would be needed. [Morrissey 1/6.37]
Assays for coagulation inhibitors
175. Various in vitro assays could be used to assess the effectiveness of a potential coagulation inhibitor. By 2001, the most commonly used assays to assess potential inhibitors of serine proteases in the coagulation cascade were clotting assays and enzymatic assays using chromogenic substrates. [Morrissey 1/6.41]
Initial testing using a chromogenic assay
176. For factor Xa inhibitors, once a starting point had been identified, the first step in the Skilled Team’s testing funnel would be to carry out in vitro chromogenic enzyme inhibition assays to assess the ability of the compound to inhibit factor Xa (and other serine proteases for selectivity) in vitro. This assay measures the ability of a factor Xa inhibitor to prevent the factor Xa-mediated cleavage of a molecule that mimics the activation site of prothrombin. When this site is cleaved in the assay, it releases a chromophore, i.e., a molecule that can be detected by a spectrophotometer. When a factor Xa inhibitor is present in the assay, less cleavage occurs and less of the chromophore is detected. By completing the assay with a number of concentrations of the inhibitor, a concentration-response curve can be produced, and parameters of potency (IC50 and Ki) can be determined for each compound tested. These assays are simple to set up (commercial kits were available for factor Xa, and other enzymes, in 2001), quick to run, and easy to control. They can be run in parallel against several different enzymes, sometimes using a high-throughput format. [Leadley 1/5.56]
177. The chromogenic inhibition assay is not influenced by the compound’s specificity (since only one enzyme is present in the assay, whereas in blood/in vivo, multiple enzymes are present). The assay is also not affected by the compound’s bioavailability, in vivo distribution, cell wall permeability or clearance rate. [Leadley 1/5.57]
Potency
178. IC50 and Ki are measures of the potency of enzyme inhibitors. [Leadley 1/5.58]
179. IC50 is the concentration of inhibitor required to reduce the enzymatic activity to half of the uninhibited value. The lower the IC50, the less of the compound is required to produce 50% inhibition and, therefore, the more potent the compound is at inhibiting enzyme activity in the assay. The IC50 value can vary since it depends on the substrate concentration used in the IC50 determination. [Leadley 1/5.58(a)]
180. Ki is the dissociation equilibrium constant of the enzyme-inhibitor complex and is used to describe the binding affinity that an inhibitor has for an enzyme. Ki is considered a more accurate measure of potency since the Ki of an enzyme-inhibitor complex is a constant and accounts for any changes in substrate concentration. [Leadley 1/5.58(b)]
181. For competitive inhibitors, IC50 and Ki are related mathematically as indicated in the following formula [Leadley 1/5.59]:
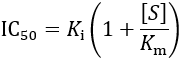
182. Where [S]=substrate concentration, Km=Michaelis constant, which is the substrate concentration at which the reaction rate is half of Vmax, Ki=inhibition constant, and IC50=the concentration of the inhibitor required to reduce the enzyme activity to half of the uninhibited value. [Leadley 1/5.60]
Selectivity
183. The terms selectivity and specificity are often used interchangeably in relation to enzyme activity. Factor Xa is a serine protease. This is a very large class/family of enzymes which include enzymes that play important roles in coagulation, fibrinolysis, digestion and inflammatory responses. [Leadley 1/5.62]
184. Other chromogenic enzyme assays would therefore be run in parallel to assess selectivity, i.e., whether a compound also inhibits other similar enzymes. The other serine proteases that the Skilled Team developing a factor Xa inhibitor would typically assess in the initial screening for selectivity for factor Xa inhibitors were trypsin, thrombin, aPC, plasmin, and tPA. Trypsin was routinely tested to examine the effect of compounds against a serine protease which is not involved in the coagulation/fibrinolysis system. aPC, plasmin, and tPA were evaluated because inhibiting these enzymes would work against the desired antithrombotic effect of a factor Xa inhibitor. The reason for these specific enzymes is that trypsin is representative of non-coagulation/fibrinolysis enzymes and aPC, plasmin, and tPA inhibition would be expected to reduce the effect of an anticoagulant. [Leadley 1/5.63]
185. A compound is considered specific or selective if there are several orders of magnitude difference in the ability of the compound to inhibit the enzyme of interest compared to other non-targeted enzymes. [Leadley 1/5.64]
Testing in clotting assays
186. In the field of anticoagulants, it was essential to assess whether compounds which may inhibit part of the coagulation cascade in an in vitro enzyme assay, in fact, resulted in an anticoagulant effect on the blood. Chromogenic enzyme inhibition assays are designed to measure the level or function of specific factors. Clotting assays provide a whole blood assessment of coagulation function and an assessment of whether a compound can effectively inhibit coagulation. Therefore, compounds which met the in vitro potency and selectivity criteria would then be selected by the Skilled Team to be tested in routine clotting time assays using laboratory animal or human blood samples. Blood would be obtained from subjects and the compound of interest would be added at several concentrations to the samples in vitro and then subjected to routine clotting assays. Two commonly used clotting assays are prothrombin time (PT) and the activated partial thromboplastin time (aPTT). [Morrissey 1/6.42; Leadley 1/5.70]
187. PT and aPTT are standard assays that measure the time it takes blood or plasma to clot after adding a clotting stimulator (thromboplastin for PT and partial thromboplastin for aPTT). This can be carried out on blood obtained from animals or on human blood. These assays are routinely used in clinical practice to monitor patients treated with warfarin or heparin. In a drug development program, a compound would be given either orally or intravenously to an animal. Blood samples are taken prior to compound administration and a specific time after compound administration. The plasma component is separated from the blood samples and each plasma sample is subjected to PT and aPTT assays. In 2001, such assays were automated using special instruments that measured clotting spectrophotometrically by detecting changes in light transmission through the sample as a clot formed. A comparison is then made between the time taken for the plasma to clot in plasma samples obtained after compound administration compared to the control sample obtained prior to compound treatment. [Leadley 1/5.71]
188. The outcome of these assays is usually measured in seconds from when an activator is added to the plasma sample until the plasma sample clots. For example, if the PT time in an untreated sample is 13 seconds and the PT time increases to 26 seconds following administration of the compound of interest, that would represent a 2-fold increase in clotting time. This would indicate that the blood is taking longer to clot, indicating that the inhibitor is having an anti-coagulant effect. The results of these assays carried out on samples of human blood were used to help translate the animal model data (described below) to human data in order to better predict the dose of compound to be tested in initial human clinical trials. [Leadley 1/5.72]
Testing for oral bioavailability
189. By 2001, a major challenge in the field of factor Xa inhibitors was that many potent and selective compounds had poor oral bioavailability. Therefore, the Skilled Team would screen compounds which met the criteria for potency and selectivity in the chromogenic assay using a preliminary test for oral bioavailability. [Leadley 1/5.66]
190. The precise preliminary test that could be used would vary, but this initial evaluation could be completed quickly and did not require a large amount of compound. The initial assessment of bioavailability could be conducted using rats that were administered doses of compounds intragastrically with doses ranging from 1 to 50 mg/kg. Arterial blood samples were then taken periodically to assess the factor Xa inhibitory activity by measuring the factor Xa inhibition activity of the sample in a chromogenic factor Xa inhibition assay. If the test compound had entered the bloodstream, the blood plasma would have greater factor Xa inhibitory activity in a chromogenic assay than blood from an untreated control. The activity of the compound in the plasma sample would be used to determine the approximate concentration of the compound in the plasma at each time point. [Leadley 1/5.67]
191. For compounds that demonstrated activity after dosing intragastrically, similar experiments would be performed with the compound administered intravenously. By comparing the concentration of drug in the plasma at various time points after intragastric dosing with concentrations achieved after intravenous dosing (which is considered to be 100% bioavailable), an estimate of oral bioavailability could be generated. [Leadley 1/5.68]
192. In addition, the intravenous administration provides data to determine the terminal half-life of the drug, which is an estimate of how long the drug stays in the blood after dosing. In rats, a favorable bioavailability result would be >40% F (“F” is the fractional absorption, comparing oral vs. intravenous administration), and a terminal half-life of > 4 hr was desirable. [Leadley 1/5.69]
Animal models
193. In general, only if a compound met the criteria for potency, selectivity, bioavailability and ex vivo activity in a clotting assay would the Skilled Team move on to test the compound for antithrombotic efficacy in an animal model of thrombosis. Several different animal models were used routinely to determine the ability of a compound to prevent formation of an experimentally-induced thrombus, either induced to form within a blood vessel or in a tube that was connected in the circulatory system. [Leadley 1/5.73]
194. Although a number of animal models of thrombosis were available for testing in vivo antithrombotic efficacy, one in vivo system that would typically be used is the rabbit arteriovenous (AV) shunt model. This is one of the standard animal models that the skilled pharmacologist would be familiar performing to assess the anticoagulant activity of compounds. A shunt or large tube is surgically placed between the carotid artery and a vein. A silk thread is placed inside the tube and as the blood flows over the thread, a clot forms on the thread. The thread is then removed and weighed and the higher the weight, the larger the clot. The baseline reading for clot formation is taken and the compound of interest is administered, and then the assay is repeated with a new shunt and thread. If the compound of interest is an effective anticoagulant it will decrease the amount of clot formed, as measured by the weight of the clot on the thread. Comparisons could also be made against known anticoagulants such as TAP or heparin. [Leadley 1/5.74]
195. Other models also used included the rat ferric chloride model. In this model, ferric chloride is placed on the exterior of a carotid artery. This damages the blood vessel and initiates thrombus formation within the vessel. After a given time, the thrombus is removed from the artery and weighed. In this model, thrombus weights from rats treated with an antithrombotic compound (administered intravenously, orally, or subcutaneously) were compared to thrombus weights from rats that were given a placebo. [Leadley 1/5.75]
Pharmacokinetics
196. Compounds that reached this stage in the testing funnel and reduced clot formation in a relevant in vivo model would be taken forward to assess pharmacokinetic (PK) parameters in detail in different animal systems, typically in rats and dogs. [Leadley 1/5.76]
197. The most important pharmacokinetic measures which were routinely assessed for factor Xa inhibitors were oral bioavailability, half-life, clearance, volume of distribution and protein binding. [Leadley 1/5.77]
198. Many potential factor Xa inhibitors which were potent and selective and reduced clot formation in animal models failed at this stage because they had poor pharmacokinetic properties, in particular poor oral bioavailability and short half-life, which would likely make them poor drug candidates. [Leadley 1/5.78]
Proteins
199. Proteins are large molecules that perform a vast array of functions within organisms including catalysing reactions, DNA replication, responding to stimuli, providing structure to cells and transporting molecules. Proteins differ from one another primarily in their sequence of amino acids, which is dictated by the nucleotide sequence of their genes. There are 20 natural proteinogenic amino acids, which each have the same basic structure, composed of a central carbon attached to a carboxylic acid, an amino group, a hydrogen atom and a variable fourth group, known as the side chain. The carboxylic acid and amino groups form peptide bonds to assemble the amino acid main chain. The side chains project from this main chain. Shorter amino acid sequences of 20-30 amino acids are often called peptides, whilst longer sequences make up proteins. The amino acids can be coded using 1 or 3 letter codes (Alanine, for example, is referred to as Ala or A) and this helps to describe the protein sequence. In addition, natural amino acids possess defined stereochemistry at the alpha-carbon atom and are commonly referred to as L or a amino acids. [Camp 1/6.11; Redshaw 1/29 & footnote 3]
200. There are four distinct levels of protein structure; primary, secondary, tertiary and quaternary (see Figure 2) below. [Camp 1/6.12]
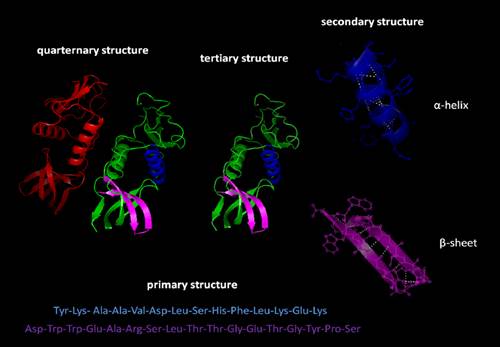
Figure 2 Four levels of protein strucutre [Camp 1/Figure 1]
201. The order in which the individual amino acids are linked together making up a protein is known as its primary structure (see Figure 3 below). [Camp 1/6.13]
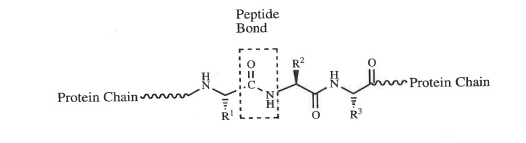
Figure 3 Primary structure of a protein [Camp 1/Figure 2]
202. An unfolded polypeptide chain is in a high energy, unfavourable state. To lower its energy, which is more favourable, the polypeptide chain will adopt a unique fold. First the polypeptide chain will fold to form areas of secondary structure. These structures are defined by patterns of hydrogen-bonds between the main-chain peptide groups. The secondary structure comprises regions of the polypeptide chain which adopt an ordered configuration. There are two main structures - the α-helix and the β-pleated sheet. An α-helix and β-pleated sheet are shown in Figure 2 above. [Camp 1/6.14]
203. These regions of secondary structure will undergo further coiling and folding to achieve a more complex structure. The overall three-dimensional (3D) structure of a protein is referred to as its tertiary structure and it is this structure of enzymes and receptors that is crucial to their function and to their interaction with drugs. Structural proteins are quite ordered in shape, whereas other proteins such as enzymes and receptors fold up on themselves to form more complex structures. [Camp 1/6.15]
204. Complexes of two or more polypeptides are called multimers and possess an additional level of structural organisation called quaternary structure. A depiction of quaternary structure is also shown in Figure 2 above. [Camp 1/6.16]
Enzymes
205. Enzymes are a class of proteins which catalyse over 5,000 biological reactions by accelerating the conversion of substrates to products. One way of achieving this is by lowering the activity energy for the reaction through stabilisation of the transition state. Without enzymes, the cell’s chemical reactions would be too slow to be useful. Enzymes act as a surface or focus for the reaction, bringing the substrate or substrates together and holding them in the best position for reaction. The reaction takes place, catalysed by the enzyme, to give products which are then released. Catalysis by an enzyme is mediated by a region of the protein termed the active site. The enzyme’s natural substrate binds at the active site and the relevant reaction is catalysed. Only substrates that bind to the enzyme active site are turned over by the enzyme. [Camp 1/6.19; Redshaw 1/29]
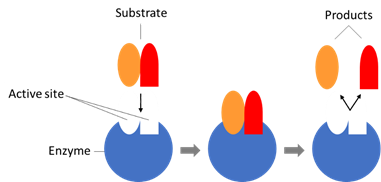
Figure 4 Protease activity [Redshaw 1/Figure 1]
206. The active site usually consists of an indentation on the surface of the enzyme that has a unique three-dimensional structure and functional group distribution. This means that enzymes are highly specific both in the reactions they catalyse and the identities of their substrates. The amino acids present in the active site play an important role in enzyme function. There are certain amino acids in the active site that remain constant between evolutionarily related enzymes. These conserved amino acids can have one of two roles: binding the substrate, co-factors or metals within the active site and/or catalysing the reaction. [Camp 1/6.20]
207. Only molecules with the right shape and functional group distribution can bind to the active site (via the formation of Van der Waal forces, electrostatic interactions, hydrogen bonding and/or hydrophobic interactions) and form the enzyme-substrate complex required for catalysis. One model for enzyme-substrate binding is the 'lock and key' model (in which both enzyme and substrate are seen as rigid with the substrate fitting like a key into a lock). [Camp 1/6.21; Redshaw 1/41]
208. Another model is the 'induced fit' model. This model assumes that the active site of an enzyme has a degree of flexibility. It proposes that the substrate is not quite the perfect shape for the active site but when it enters the active site the latter changes shape slightly to maximise bonding interactions. [Camp 1/6.22]
209. Enzymes that have a substrate in common will all have a space in their active site that is complementary to the three-dimensional shape and chemistry of the functional groups of the relevant substrate into which the relevant substrate can bind. [Camp 1/6.23]
210. Enzymes may require the presence of cofactors to carry out the catalysis. Cofactors are non-protein substances that are required for the activity of the enzyme. They can be metal ions (zinc or iron, for example) or small organic molecules called co-enzymes. [Camp 1/6.24]
211. The activity of most enzymes is tightly regulated in the body. For example, enzymes can be regulated by molecules which bind to allosteric sites on the enzyme (i.e. a binding site distal from the active site of the enzyme). Enzymes can be regulated by other enzymes. For example, the protein kinases phosphorylate specific residues on target enzymes, leading to their activation or deactivation. [Camp 1/6.25]
212. Enzymes are not static, rigid structures, but are flexible and operate through an ensemble of conformational changes. Amino acid residues, protein loops and secondary structures can move and adopt different states, thus altering the functional activity of the enzyme. [Camp 1/6.26]
Serine Proteases
213. Since enzymes catalyse a range of different reactions, they can have very different structures. Enzymes are first grouped according to their function; for example proteases, which cleave peptide bonds, phosphatases, which remove phosphate groups, and methyltransferases, which transfer methyl groups. [Redshaw 1/30]
214. There are four major classes of protease enzymes (aspartyl, serine, cysteine and metallo) that selectively catalyse the hydrolysis of peptide bonds. The catalytic mechanism is different for each class and typically involves nucleophilic attack on the scissile amide bond of the substrate (a covalent bond that can be broken by an enzyme) by a specific amino acid residue of the enzyme or activated water molecule. For example, serine proteases employ an active site nucleophilic serine residue to mediate the cleavage of the peptide bond, aspartyl proteases have an aspartic acid residue in the active site, and metalloproteases have a metal ion in the active site. Within a given mechanistic family, the catalytic apparatus is very similar but there are likely to be other differences at the active site so that different natural substrates can bind. [Camp 1/6.34; Redshaw 1/30]
215. Serine proteases can be categorised based upon the substrate specificity of the enzymes to include trypsin-like, elastase-like and thrombin-like. Factor Xa is a trypsin-like serine protease which cleaves peptide bonds adjacent to positively charged residues such as arginine. Serine proteases employ a catalytic triad in their active sites involving three key amino acid residues: aspartic acid (Asp), histidine (His) and serine (Ser). Binding of the substrate in the active site triggers a series of events that lead to peptide cleavage. [Camp 1/6.35]
216. All proteases have a number of ‘binding pockets’ in the vicinity of the active site in which the substrate binds. Most proteases are sequence specific; the size and the hydrophobic/hydrophilic character of their binding pockets determines their substrate specificity. For instance, a polar aspartic acid residue within a protease binding pocket would be ideally suited to interact with the basic side chain of a suitably positioned arginine within a substrate. [Redshaw 1/31; Camp 1/6.36]
217. There is a commonly used convention for describing the binding pockets and the corresponding substrate amino acids. In the substrate, starting from the peptide bond that is cleaved (the scissile bond), N-terminal amino acids are termed P1, P2, P3 etc., whilst C-terminal amino acids are termed P1’, P2’, P3’ etc. These amino acids bind into pockets in the protease that are termed S1, S2, S3 etc. or S1’, S2’, S3’ etc., respectively, as shown in Figure 5 below (i.e. P1 of the substrate binds into S1 of the protease etc.). [Redshaw 1/31; Camp 1/6.36]
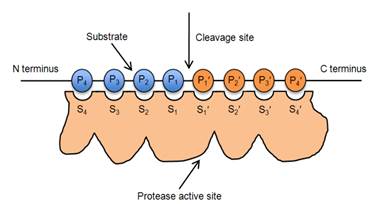
Figure 5 Nomenclature of protease substrate and binding pockets [Redshaw 1/Figure 2]
Enzyme Inhibitors
218. Molecules that interact with an enzyme and reduce its activity by influencing the binding of substrate and/or the number of reactions the enzyme turns over per unit time are known as inhibitors. There are various mechanisms through which enzyme inhibitors can act. [Camp 1/6.27; Redshaw 1/32]
Competitive (reversible) inhibitors
219. The binding interactions between substrate and enzyme have to be properly balanced so that they are strong enough to hold the substrate(s) at the active site of the enzyme to allow the reaction to take place but are weak enough to allow the products to leave (otherwise the enzyme would become “clogged up”). A molecule that binds to the enzymatic binding site, thus competing directly with a normal substrate for an enzymatic binding site, can function as a competitive inhibitor. A competitive inhibitor usually bears some features of the substrate to the extent that it specifically binds to the active site but differs from the substrate enough to be chemically unreactive (or react very slowly). The effect of a competitive inhibitor is reversed by increasing the concentration of substrate because the frequency of successful collisions between inhibitor and active site is reduced. A competitive inhibitor therefore acts by reducing the concentration of free enzyme available for substrate binding. [Camp 1/6.28; Redshaw 1/32]
220. A general model for competitive inhibition is given by the following scheme, in which the element denoted “DRUG” is shown as a competitive inhibitor for the substrate [Camp 1/6.29]:
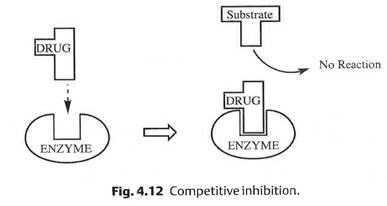
Figure 6 General model for compeititve inhibition [Camp 1/Figure 4]
Non-competitive, reversible (allosteric) inhibitors
221. These compounds bind into an allosteric site (i.e. a binding site distal from the active site of the enzyme) which changes the affinity of the enzyme for its substrate by triggering a change in the 3D shape of the active site. The inhibitor may bind reversibly, in which case the active site of the enzyme will return to the correct 3D shape for catalysis. Many enzymes are regulated naturally by allostery. [Camp 1/6.30; Redshaw 1/32]
Figure 7 General model for non-competitive, reversible inhibition [Camp 1/Figure 5]
Non-competitive (irreversible) inhibitors
222. An irreversible inhibitor binds irreversibly to the active site (via, for example, the formation of a covalent bond) and permanently blocks substrate from binding. Irreversible inhibitors typically bind in the active site rather than allosterically, since the amino acids responsible for catalysis are those that covalently bind to the inhibitor. [Camp 1/6.31; Redshaw 1/33]
Figure 8 General model for non-competitive, irreversible inhibition [Camp 1/Figure 6]
223. In 2001, reversible inhibitors were generally preferred. This was because irreversible, covalently-bound inhibitors raised safety concerns for two reasons. Firstly, there were concerns that irreversibly inhibiting the enzyme itself could have unwanted effects since new protein synthesis would be needed to restore enzymatic activity. Secondly, the reactivity of groups needed for irreversible inhibition might mean that the inhibitor would bind to other, unrelated, proteins, leading to more frequent unwanted off-target effects. [Redshaw 1/34]
Potency
224. The ability of an inhibitor to inhibit the activity of an enzyme can be measured, and is termed ‘potency’. In general, the more tightly an inhibitor binds to the active site, the more ‘potent’ it is said to be and, the more potent the inhibitor, the less of that compound that is needed to achieve a given level of inhibition. The potency of an enzyme inhibitor in vitro is typically expressed as either an IC50 or Ki value. Although both values provide a good indication of inhibitor potency, they are not identical. The IC50 is the concentration of inhibitor required to reduce enzymatic activity by 50%. For a competitive reversible inhibitor, the IC50 depends on the concentration of the substrate since the inhibitor and substrate compete for the same active site. The Ki is the dissociation constant describing the binding affinity between the inhibitor and the enzyme and is not dependent on substrate concentration. Since both values refer to concentrations, compounds with a lower Ki or IC50 are more potent. For a given inhibitor against a given enzyme, the Ki will be slightly lower than the IC50. [Redshaw 1/36; Camp 1/6.40]
Selectivity
225. For an enzyme inhibitor to be a useful drug almost always requires that it is selective (also referred to as ‘specific’), which means that it is many fold more potent against the enzyme of interest, compared with other enzymes in the body. This is because if the compound inhibits another enzyme there can be unwanted side effects, which in turn may mean that a compound is unsuitable for therapeutic use. [Redshaw 1/37]
226. The skilled medicinal chemist would also consider selectivity over other proteins in the body more broadly. There are unrelated ‘off target’ proteins that the skilled medicinal chemist will almost always look to avoid. An example here is cytochrome P450, an important metabolising enzyme in the liver. Inhibition or induction of cytochrome P450 can lead to drug-drug and drug-food interactions and this would have been something that the skilled team would have wished to avoid. [Redshaw 1/38]
Enzyme Kinetic Studies
227. Enzyme kinetics is the study of the rate at which enzymes catalyse their specific chemical reactions. The rate of catalysis depends on the concentration of enzyme, substrate, and inhibitor present, as well as factors such as pH and temperature. The binding affinity of an inhibitor and the binding affinity of a substrate can all be determined empirically through enzyme kinetic studies. Whilst the skilled medicinal chemist would look at and interpret the results of these enzyme kinetic studies, he would generally not carry out the tests himself. [Camp 1/6.32]
Drug-Like Properties
228. It is often the case that potent and selective in vitro activity does not translate into good in vivo activity. For good activity in vivo, a compound which is intended to be administered orally must also possess other ‘drug-like’ properties, including sufficient solubility, permeability, oral bioavailability and half-life. Drug attrition and high failure rates for clinical candidates had been a major concern of the pharmaceutical industry for many years. There are many reasons a clinical compound could fail to become a new medicine, including lack of connection of the biological target to the human disease, poor pharmacokinetics or toxicity. [Redshaw 1/39; Camp 1/6.45]
Potent and Selective Enzyme Inhibitors
229. Part of the skilled medicinal chemist’s role is to design and synthesise molecules that could be potent and selective inhibitors of the target enzyme (here factor Xa). To do this, the skilled medicinal chemist has in mind the intermolecular interactions that contribute to the binding of small molecules to the enzyme’s active site. These include both enthalpic and entropic contributions. That said, there are a wide variety of these interactions and the interplay between them can be complex. Indeed, minor modifications to a compound’s structure can result in major and unpredictable changes in binding activity. [Redshaw 1/40]
230. The features that the skilled medicinal chemist would focus on include: [Redshaw 1/41]
(a) conformation (shape) of inhibitor: the conformation of the inhibitor is the 3D shape that it will typically adopt in solution. Pre-shaping the inhibitor into a rigid conformation to match the shape of the active site of the target enzyme leads to smaller loss in conformational entropy on binding (making for stronger binding). This could also lead to improved selectivity. On the other hand, if a rigid conformation does not match the shape of the active site, the strength of binding is reduced and can even be lost; [Redshaw 1/41.1; Camp 1/6.21]
(b) steric effects: two atoms cannot occupy the same space, and when two atoms (or groups of atoms, as in a compound) are forced together, there is an unfavourable interaction termed a steric clash or steric hindrance. In the case of an enzyme inhibitor, even introducing a small group, such as a methyl (-CH3) group, can result in a significant loss in potency if a steric clash occurs and the compound can no longer fit snugly into the active site; [Redshaw 1/41.2]
(c) hydrogen bonds: these are primarily electrostatic forces that arise between a hydrogen atom and an electronegative atom or group, either in the same or a different molecule (see Figure 9 below). These bonds are often quite strong (although the strength can vary considerably depending on e.g. the alignment of the three atoms making up the bond), and so the skilled medicinal chemist will look to try and design compounds that could potentially form hydrogen bonds with amino acids around the active site of the enzyme; [Redshaw 1/41.3; Camp 1/6.18(c)]
Figure 9 Hydrogen bond (shown in red) between serine residue side chain (bottom) and main chain carbonyl group (top) [Redshaw 1/Figure 3]
(d) salt bridges/ionic bonds: these are electrostatic interactions formed between two fully charged groups with opposing charges (see Figure 10). In the context of proteins, negatively charged aspartate and glutamate residues (formed by deprotonation of aspartic acid or glutamic acid side chains) can each form salt bridges with positively charged species such as ammonium or guanidinium ions (formed by protonation of lysine or arginine side chains). Such interactions can be significantly stronger than hydrogen bonds between uncharged species; [Redshaw 1/41.4; Camp 1/6.18(a)]
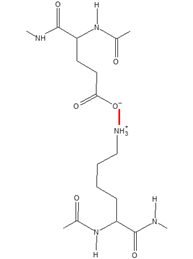
Figure 10 Salt bridge (shown in red) formed between ammonium ion (protonated lysine side chain residue, bottom) and glutamate (deprotonated glutamic acid side chain residue, top) [Redshaw 1/Figure 4]
(e) Permanent dipole-dipole interactions - a permanent dipole is a permanently uneven distribution of electronic charge between atoms of different electronegativities bonded together. The atom of higher electronegativity pulls electron density towards itself resulting in partial positive and negative charges within the molecule (i.e. a polar molecule). Electrostatic forces of attraction can form between oppositely partially charged groups of two dipoles. [Camp 1/6.18(b); Redshaw 1/42]
(f) Van der Waals interactions - take place between hydrophobic molecules (for example between aromatic groups or between aliphatic groups). These arise from the fact that the electronic distribution in neutral residues is never totally symmetrical. There are always transient areas of high electron density and low electron density such that an area of high electron density on one residue can have an attraction for an area of low electron density on another molecule. [Camp 1/6.18(d); Redshaw 1/42]
(g) π interactions - these are interactions based on the increased electron density present in systems with delocalised π electrons such as multiple C=C bonds in an aromatic ring. These delocalised electrons can interact with neighbouring species such as another π system, a polar molecule, a cation, an anion, and a metal (cationic or neutral). [Camp 1/6.18(e); Redshaw 2/footnote 18]
(h) Hydrophobic interactions - in an aqueous environment, it is more energetically stable for non-polar groups to group together rather than remain dispersed throughout the water molecules. Water molecules are polar (i.e. contain areas of positive and negative charge). In the presence of a non-polar group, the surrounding water molecules prefer to interact with each other and form an ordered cage-like structure, therefore existing in a higher (unfavourable) energy state. If non-polar groups come together, this reduces the number of water cages and releases 'free' water molecules, thereby lowering the overall energy. [Camp 1/6.18(f); Redshaw 1/42]
Drug Discovery Process
N.B. The precise wording below is taken from Dr Redshaw’s First Expert Report. Whilst there is general agreement between the medicinal chemists at a high level, it may be useful to also read Dr Camp’s overview of the drug discovery process at paragraphs 6.37 to 6.44 of his First Expert Report.
231. Research into new drugs is a trial and error process, which means that drug discovery is difficult, expensive and time-consuming. In 2001, it would typically take a minimum of 2–3 years to go from the start of a project to a clinical candidate, although the vast majority of projects would not deliver a clinical candidate. Even then, of these candidates the vast majority would fail in clinical trials, which themselves can take up to a further 10 years. It would not be unusual for a medicinal chemist to spend their entire 30–40 year career synthesising compounds without ever having worked on a marketed drug. [Redshaw 1/43]
232. In 2001, the drug discovery process typically encompassed a number of stages: [Redshaw 1/44]
(a) The first stage, often referred to as “Target Discovery” (or “Target Identification”), involves the identification of a biological target or pathway that could potentially play a role in the disease. In the case of thrombosis, it was hypothesized that intervention within the blood coagulation pathway could lead to anticoagulant medicines. Early research identified thrombin as an initial target of interest and later factor Xa emerged as an additional target for intervention. [Redshaw 1/46; Camp 1/6.38]
(b) it would be necessary to identify a compound (or class of compounds) that might be promising for further development based on in vitro potency and possibly selectivity against any key off-target proteins. Preliminary results from in vitro DMPK assays could also be used if the data were available; [Redshaw 1/44.1; Camp 1/6.39]
(c) if a promising compound/class of compounds was identified, the skilled medicinal chemist would then synthesise variations around the compound(s) and test their potency, selectivity and DMPK properties in the hope that it would be possible to build up an idea of SAR (Structure-Activity Relationships). [Redshaw 1/44.2; Camp 1/6.40]
(d) using the SAR to narrow the choice of potential compounds to synthesise, the skilled team would synthesise and test compounds in the hope of identifying molecules with sufficiently good potency, selectivity and in vitro DMPK properties to be potentially useful as a drug; [Redshaw 1/44.4; Camp 1/6.41]
(e) the most promising compounds would then be selected for in vivo testing. These tests would include in vivo DMPK studies and efficacy in an animal model of disease; [Redshaw 1/44.4]
(f) the most promising compounds from the in vivo testing (if any) would then move on for further DMPK and toxicology screening; and [Redshaw 1/44.5]
(g) if any compound was predicted to be sufficiently safe and effective in humans, the drug candidate would enter into clinical trials. [Redshaw 1/44.6; Camp 1/6.41]
Starting point
233. The skilled medicinal chemist would often first become involved in a new drug discovery project once the biological target had been identified. Their first task would be to work with the skilled pharmacologist to identify a compound (or class of compounds) as a starting point for the project (in this case potential inhibitors). The objective would be to find a compound or compounds with sufficient activity against the target, and possibly selectivity for that target over other enzymes, which could form a starting point for synthesising analogues, which may have improved properties. Starting points can typically be identified in three different ways: i) finding known compounds published in the literature; ii) starting from the endogenous substrate[7]; and iii) high-throughput screening, which was already in use in 2001, and in which tens or hundreds of thousands of compounds can be screened using automated assays. [Redshaw 1/46; Camp 1/6.39]
234. In a well-developed field where there were a significant number of known compounds with good activity at the target (and potentially other desirable drug-like properties), the skilled medicinal chemist would most likely identify the most promising compounds for further work based on known compounds published in the literature. If this proved unsuccessful, other approaches may also have been tried, for example high-throughput screening. [Redshaw 1/47]
Optimisation
235. Once a starting point had been identified, the skilled medicinal chemist would most likely adopt an iterative approach to modify the structure(s) of the starting compound(s) to try to improve activity, selectivity and other drug-like properties. This is done by designing different structural analogues of the starting compound(s), keeping in mind the potential impact of structural modifications on all of the interactions described above. In order to rationalise the design process, medicinal chemists typically build up SAR of the compounds being optimised. Computer modelling can also be used. [Redshaw 1/48; Camp 1/6.40]
236. There are various strategies which can be used to improve the interactions between a drug and its target, including (i) variation of substituents; (ii) extension of the structure; (iii) chain extensions/contractions; (iv) ring expansions/contractions; (v) ring variations; (vi) ring fusions; (vii) isosteres; (viii) simplification of the structure; and (ix) rigidification of the structure. [Redshaw 2/26]
237. One strategy the skilled medicinal chemist could try in an attempt to improve the activity of a given compound (which would result in the formation of a new compound with a different chemical structure to that of the original compound) and reduce its side-effects is “rigidification”. One well-known method of rigidifying a flexible molecule is to incorporate the skeleton of the flexible molecule into a ring system (cyclisation). Flexible side chains can also be rigidified by incorporating a rigid functional group e.g., a double bond, alkyne, amide or aromatic ring. [Camp 1/6.50; Redshaw 2/26]
238. Less flexible inhibitors can result in stronger binding, and cyclisation was a common way of making less flexible compounds in 2001. However, the outcome will depend on whether the conformation that the compound is ‘locked’ into is one that is favoured for binding. If it is, rigidification will increase the strength of binding; the reduction in the number of conformations may also reduce interactions with other enzymes or receptors and hence improve selectivity. However, rigidifying an inhibitor could also have the opposite effect, since the inhibitor could be locked in a conformation that is less favourable for binding, thus reducing binding affinity or even eliminating binding altogether. [Redshaw 2/28]
239. The drug optimisation process is uncertain and there is no guarantee of success with any of the optimisation strategies listed above. [Redshaw 2/30]
Structure-Activity Relationships
240. As noted above, given the difficulty in predicting potency and selectivity based on structure alone, the skilled medicinal chemist would approach optimisation rationally by trying to generate SAR. To do this, the skilled medicinal chemist would make a series of small structural modifications to the initial compound, resulting in a number of different structural analogues. These analogues would be tested to determine how each of the modifications affects activity against the target. Often relatively small modifications can result in significant changes in activity against the target enzyme or against other enzymes. By measuring the activity of each analogue against the target enzyme (and its selectivity by measuring activity against other enzymes), the skilled medicinal chemist builds up an idea of which parts of the compounds are important for binding, and the size and nature of each of those parts required for strong binding. However, this is very much an empirical process as it can take a large amount of trial and error experimentation. [Redshaw 1/49]
241. SAR optimisation to improve potency and selectivity would typically be conducted by a medicinal chemist, working with a pharmacologist/biologist. By 2001, however, SAR optimisation work in pharmaceutical companies typically also monitored other drug properties (e.g. bioavailability, metabolism) in parallel with work to improve potency and selectivity, and would therefore also involve a DMPK scientist. [Redshaw 1/50; Camp 1/6.41]
Crystal structures/computer modelling
242. A medicinal chemist could supplement their SAR work using computer modelling and, if available, crystal structures. A crystal structure, which provides a 3D picture of the enzyme active site and the amino acids within it, is obtained by growing a single crystal of the target protein, and then using an X-ray to obtain a diffraction pattern. A crystallographer, with the aid of computer programs, can then calculate the structure of the protein from the diffraction pattern. This can be done with either the native enzyme (without a bound inhibitor) or, more usefully, with a bound inhibitor. [Redshaw 1/51; Camp 1/6.40]
243. In 2001, growing protein crystals could be a difficult and laborious process (depending on the protein). It was, indeed, not uncommon that a crystal structure was not produced until after a drug candidate had been identified, when it was useful to demonstrate how the drug candidate binds to the target. If the crystal structure of the target was not available, computational scientists could often develop a homology model based on the crystal structure of a similar protein. However, where a crystal structure was available, knowledge of the binding site and how a molecule binds could be helpful in identifying structural modifications that could potentially improve binding (and therefore potency). [Redshaw 1/52; Camp 1/6.40]
Factor Xa inhibitors
State of the Field
244. In 2001, a number of pharmaceutical companies were reported to be developing factor Xa inhibitors. There were a large number of compounds that had been found to be very potent factor Xa inhibitors with Ki or IC50 values in the nanomolar (and even sub 1 nM) range. Their selectivity with respect to thrombin and trypsin was often reported to be high and, for some compounds, in vivo data from animal models were described. An orally bioavailable factor Xa inhibitor would be much preferred over one that had to be delivered parenterally. [Redshaw 1/58; Camp 1/6.63, 6.66, 6.67 & 6.68]
Factor Xa structure
245. The crystal structure of human factor Xa had been published in 1993. Subsequently, crystal structures with bound inhibitors (including DX-9065a - see below) were also published, showing how synthetic small molecule inhibitors bind to the factor Xa binding pockets. [Redshaw 1/59; Camp 1/6.53 & 6.74]
246. Proteases have at their active sites a number of specificity pockets (S1, S2, etc.) into which the substrate binds. The crystal structures of factor Xa bound to different inhibitors showed that the key binding pockets for small molecules binding to factor Xa are S1 and S4, which were well characterised. S1 is a deep, narrow pocket with hydrophobic walls and an aspartic acid (Asp189) at its base. The S4 pocket has distinct sub-regions; a ‘hydrophobic box’ and a negatively charged cation binding hole. [Redshaw 1/59; Camp 1/6.59 & 6.60]
Selectivity
247. Factor Xa, trypsin and thrombin all belong to the trypsin-like family of serine proteases, which cleave after a basic amino acid (i.e. the natural substrate has a lysine or arginine at P1). The skilled medicinal chemist would be aware that an inhibitor of factor Xa may also inhibit these proteases because of their similar binding pockets. In 2001, it was known that there were subtle differences in binding pockets between factor Xa, trypsin, and thrombin. Such differences could potentially be exploited to provide selective inhibition of these proteases i.e. by maintaining binding at factor Xa while reducing binding at trypsin and thrombin. Further, it was also important to have selectivity over a larger panel of other serine proteases, including aPC, plasmin and tPA. [Redshaw 1/61 & 62; Camp 1/6.40, 6.57 & 6.59]
Overview of factor Xa inhibitors in development at the priority date
248. By 2001, the pharmaceutical industry had developed and analysed a large and diverse group of potential inhibitors. [Redshaw 1/63]
249. One of the first published lead compounds was DX-9065a , developed by Daiichi. DX-9065a was reported to be potent and selective, to have some oral bioavailability and to have efficacy in animal models. DX 9065a had also been progressed into a preliminary clinical study. [Redshaw 1/66; Camp 1/6.73, 6.74 & 6.75]
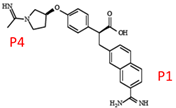
Figure 11 Annotated structure of DX-9065a [Redshaw 1/Figure 5]
250. A crystal structure of DX-9065a bound to factor Xa had been generated and published. This showed the benzamidine group (labelled as P1 in the figure above) binding to the aspartate residue at the bottom of the S1 pocket of factor Xa and the other amidine group (labelled as P4 in the figure above) binding to the cation hole in the S4 pocket. This crystal structure also showed that DX-9065a adopts an L-shaped conformation when bound, which the literature reports to be necessary to place the P1 and P4 groups in their respective binding pockets, i.e. pockets S1 and S4, in factor Xa. This relative orientation of the P1 and P4 groups in a L-shaped conformation linked by a central “core” was thought to be important for potent inhibition. [Redshaw 1/67; Camp 1/6.62, 6.80 & 6.81]
251. In terms of the chemical groups binding in the S1 and S4 pockets of factor Xa, the amidines at the P1 and P4 positions of DX-9065a are highly basic. Highly basic compounds typically permeate through the gut wall and into the blood stream more slowly than less basic or neutral compounds. This means less drug is absorbed, resulting in lower oral bioavailability. [Redshaw 1/68; Camp 1/6.49]
252. Although DX-9065a is a potent and selective factor Xa inhibitor, its low oral bioavailability meant it was not an ideal candidate as an oral drug. A number of other compounds that use basic P1 and P4 groups are also reported to be potent and selective, but would be expected to have the same issue of poor oral bioavailability. Efforts to improve oral bioavailability appear to have focussed on replacing the amidine groups by less basic or neutral groups. [Redshaw 1/69; Camp 1/6.81 & 6.97]
253. The skilled team would also have been aware of at least the following inhibitors:
a) RPR-120844 (Rhone Poulenc Rorer) [Leadley 1.5.86(c); Camp 1/6.76]
b) Betz compound 6 (DuPont) [Camp 1/6.77; Redshaw 1/Table 1, row 2]
c) ZK-807834 (Berlex Bioscience) [Leadley 1/5.86(a); Camp 1/6.78 - 6.80; Redshaw 1/Table 1, row 9]
d) DPC-423 (DuPont) [Leadley 1/5.86(d); Camp 1/6.84 - 6.86; Redshaw 1/Table 1, row 11]
e) RPR-208815 (Rhone Poulenc Rorer) [Leadley 1/5.86(c); Camp 1/6.83; Redshaw 1/Table 1, row 4]
Binding Modes
254. An interesting feature of factor Xa inhibitors reported by 2001 is that the binding orientation of inhibitors can ‘flip’, so that the group intended to bind in the S1 pocket actually binds in the S4 pocket, and vice versa. [Redshaw 1/70 & 71; Camp 1/6.74 & 6.90]
The below is without prejudice to the Claimants’ position that WO 652 is not addressed to the skilled DMPK scientist.
Pharmacokinetics (PK) / ADME
255. Pharmacokinetics may be thought of as the study of the effects of the body on a drug. In many cases, drug therapy involves administering a dose of a medicine that is intended to be delivered to the patient’s bloodstream in order for the drug to elicit its pharmacologic activity. Blood is comprised of both cellular (red blood cells, white blood cells, platelets) and liquid components. The liquid portion of blood is called plasma. Besides water, plasma contains proteins, enzymes, antibodies, clotting factors and other components. [Taft 1/7.6]
The Desirable Properties of Drugs
256. Developing a drug is an arduous, expensive and lengthy process. Not only must the compound be potent at its target, but it must also: i) reach and stay at that target after administration in a sufficient concentration to exert a therapeutic effect; ii) avoid being metabolised and eliminated too quickly; and iii) avoid interacting with multiple other proteins. Obtaining a compound with all the required properties to navigate these issues is a careful balancing act. [Read 1/26; Taft 1/7.6 & 7.20]
257. The key DMPK properties that the skilled DMPK scientist would be interested in are: i) those that influence the four key planks of DMPK work - absorption, distribution, metabolism and excretion (“ADME”); and ii) the difference in the plasma concentration required for a drug to be effective as compared to the plasma concentration that leads to adverse side effects and toxicity. For most drugs, the aim of an appropriate dosing regimen is to maintain the drug concentration at the level required for efficacy, but below the level at which toxicity becomes a problem. [Read 1/27; Taft 1/7.7 & 7.18-7.19]
Absorption
258. A drug must reach its site of action for it to have any therapeutic effect. Absorption through the gastrointestinal tract (the “A” in ADME) is not a relevant consideration for drugs that are administered directly into the bloodstream (e.g., intravenously). However, for a compound to reach the blood plasma following oral administration requires the compound to be absorbed, survive first pass metabolism at the gut wall and the liver, and enter into the systemic circulation. [Read 1/29; Taft 1/7.8-7.9]
259. The ability for a compound to enter into the systemic circulation following oral administration is often measured as oral bioavailability (abbreviated to “F”) and presented as a percentage. F of 100% means that all the compound administered reaches the systemic circulation, and 0% means none of the compound administered reaches the systemic circulation. To calculate F, a dose is administered intravenously (“IV”) and plasma samples are collected to work out the amount of compound present in the plasma over a predetermined time (calculated as the area under the curve “AUC”). It is assumed that IV administration results in 100% bioavailability given all the compound is placed immediately into the blood. The same dose is then administered orally and the same data collected. F is then calculated using the following formula: [Read 1/30; Taft 1/7.10]
260. For a compound which is administered orally to achieve a high bioavailability requires overcoming a series of hurdles:
(a) Firstly, a compound must be in aqueous solution in order to be absorbed. This requires a compound to be hydrophilic enough to be soluble in water so that it dissolves when ingested. [Read 1/31.1; Taft 1/7.10]
(b) Secondly, the compound must be able to tolerate the extremely acidic conditions in the stomach, where the pH can be as low as 1.5. Some chemical groups may be liable to decompose under such conditions. [Read 1/31.2]
(c) Thirdly, the compound must be able to pass through the gut wall into the blood in the hepatic portal vein. This is most often achieved via passive transcellular permeability. This requires the compound to be lipophilic enough to pass through the cell membrane in the intestine, whilst still being hydrophilic enough to be water soluble. The lipophilicity of a compound is measured using log P or log D , where a higher value indicates greater lipophilicity. The lipophilicity of a compound is inversely related to its polarity (i.e. the more polar a compound is, the less lipophilic (and hydrophobic) it is). [Read 1/31.3; Taft 1/7.10]
(d) Fourthly, the compound must survive first pass metabolism in the gut wall and liver before it reaches the systemic circulation (the compound may also face metabolism in the blood plasma). [Read 1/31.4; Taft 1/7.10]
261. In 2001, standard in vitro assays were available to determine solubility, and chemical stability at different pHs, and to provide an indication of gut wall permeability (including Caco-2 and MDCK monolayer assays). [Read 1/31.1-31.3]
262. A schematic depicting the absorption of a drug administered in an oral dosage form (like a tablet) is provided below in Figure 12, which also shows the drug’s movement from administration to the point where it enters the bloodstream (i.e., the systemic circulation). [Taft 1/7.9]
Figure 12 Schematic overview of oral drug absorption. Bioavailability (F) reflects the fraction of the oral dose of drug (in blue) that is delivered to the systemic circulation [Taft 1/Figure 1]
263. The dependence of bioavailability on these processes can be described by the following equation: [Taft 1/7.10; Read 1/31]
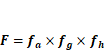
264. In the equation, fa represents the fraction of the dose absorbed into the intestine, fg is the fraction of dose absorbed into the intestine that escapes intestinal metabolism, and fh is the fraction of dose absorbed into the bloodstream that escapes “first pass” hepatic metabolism. The equation demonstrates that the overall bioavailability of a drug that is orally administered is the product of the fraction of drug that escapes loss in each organ (stomach, intestine, liver). [Taft 1/7.11]
265. The rate of drug absorption can be influenced by a number of factors including the physicochemical properties of the drug, the nature of the drug delivery system, physiologic factors, environmental factors, and the presence of underlying disease. Since drug absorption following oral administration occurs primarily in the small intestine, the rate of absorption also depends on gastric emptying time. Gastric emptying time is the time it takes for the stomach contents to empty into the intestine. A number of variables (e.g., exercise, body position, medications) affect this gastric emptying process, but perhaps the most important determinant of gastric emptying is food. [Taft 1/7.12]
Distribution
266. Once a compound has entered the systemic circulation, it can then be retained within the blood, or distributed into the body’s tissues (the “D” in ADME). How widely the compound is distributed is measured as the volume of distribution (“VD”). A low VD indicates that the compound is retained within the blood, and a high VD means extensive distribution into tissue. [Read 1/32; Taft 1/7.13]
267. VD (in liters, L) is a proportionality constant relating the amount of drug in the body as compared to its concentration in the plasma, as defined by the following equation: [Taft 1/7.15 & 1/7.26(a)]
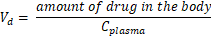
268. VD is considered an apparent volume of distribution because it does not represent an actual volume. Although the total amount of water in a person’s body is approximately 42 L, some medications have VD values in excess of 1000 L. [Taft 1/7.26(a)]
269. The larger the VD, the greater the distribution throughout the body and the lower the plasma concentration for a given dose. The dose needed to produce target plasma concentrations (e.g., Cmax) depends on VD. Drugs with larger VD will distribute more into tissues, with less drug remaining in the plasma. This will require greater doses to achieve target plasma concentrations. [Taft 1/7.26(a)]
270. As with F, determining VD requires in vivo experiments. Key factors which affect VD are: [Read 1/32; Taft 1/7.26(a)]
(a) the extent to which a compound is bound to plasma proteins. Compounds which are highly and tightly bound to plasma proteins will be retained more within the blood. Plasma protein binding has an effect on the efficacy of a drug, as it is only the unbound or ‘free’ fraction of a drug that can act at its target. In vitro assays for estimating plasma protein binding were available in 2001; [Read 1/32.1; Taft 1/7.14 & 1/7.26(a)]
(b) how hydrophilic or lipophilic a compound is. Compounds which are hydrophilic tend to have a low VD and are therefore more likely to be retained in the blood, whereas lipophilic compounds generally have a higher VD and are therefore likely to be more widely distributed into tissue; and how acidic or basic a compound is. Compounds which are basic tend to have a higher VD. [Read 1/32.2-32.3; Taft 1/7.26(a)]
Metabolism and Excretion
271. Once in the systemic circulation, a compound then needs to remain in the blood for an appropriate period of time at a high enough concentration to keep exerting its therapeutic effect. The compound will be undergoing metabolism and excretion (together referred to as ‘elimination’) such that its plasma concentration is continuously dropping. There are two main clearing organs in the body: the liver and kidney. The liver is primarily responsible for drug metabolism (the “M” in ADME). Metabolism is the process by which the body modifies the molecular structure of the drug, usually resulting in a form that is inactive or more easily removed from the body (in the form of metabolites). The kidney is primarily responsible for excretion (the “E” in ADME). Excretion is the process by which the drug and/or its metabolites are physically removed from the body, usually either in the bile or in urine. [Read 1/33; Taft 1/7.16 & 7.17]
272. The rate of elimination is shown by the drug’s half-life, which is the time taken to reduce the plasma concentration by half. The longer the t1/2, the longer it takes for the drug to be eliminated from the body. [Read 1/33; Taft 1/7.26(c)]
273. A drug’s t1/2 depends on the drug’s VD and Cl (two independent pharmacokinetic parameters) according to the following equation. [Taft 1/7.26(c)]
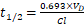
274. The equation above can be rearranged into the following equation, and this shows that the t1/2 directly depends on the relative magnitude of VD and Cl: [Taft 1/7.26(c)]
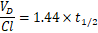
275. When administered orally, the concentration of a compound in the blood plasma is usually a curve as shown in Figure 13 below. [Read 1/33]
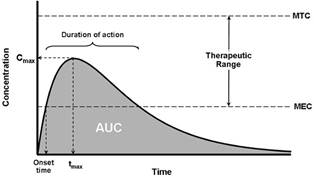
Figure 13 Oral drug concentration time curve [Read 1/Figure 2]
276. Cmax is the maximum concentration reached in the blood-stream, and tmax is the time taken to reach this concentration after administration. The area under the curve (“AUC”) is a measure of how much drug is exposed to the body (commonly referred to as systemic exposure). AUC is typically calculated from time 0 (when the dose is administered) to the time when the dose is completely eliminated from the body. The Cmax must fall under the maximum tolerated concentration or dose (“MTC”) to avoid toxicity (e.g. for anti-coagulants, excessive bleeding). When the concentration drops below the minimum effective concentration (“MEC”), the patient needs to take another dose to ensure the concentration remains within the therapeutic range. Ideally, a drug would only need to be taken once or at most twice daily, which helps maximise patient compliance with the treatment regimen. Therefore, the half-life should reflect that (e.g. depending on the therapeutic range, the half-life should be around 12-48 hours for a once-daily regimen). Figure 14 below illustrates how plasma concentration might typically vary during repeated administration: [Read 1/34; Taft 1/7.23-7.25 & 7.27]
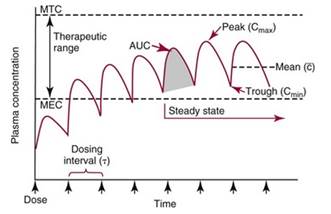
Figure 14 Drug concentration during repeated administration [Read 1/Figure 2]
277. Figure 19 shows that with repeated administration the plasma concentration has a peak and a trough. Further, it is preferable for the peaks and troughs at steady state to be shallow such that they are well within the boundaries of the therapeutic range. This can be aided by slower absorption, which reduces Cmax, and/or a longer half-life, which allows the desired dosage regimen to be met. [Read 1/35; Taft 1/7.25 & 7.27]
278. Cmax, MTC, MEC and elimination half-life can only be determined using in vivo experiments, and these values are typically species-specific. Therefore while animal models are used to predict these values in humans, these measurements are only firmly established in humans during clinical studies in late-stage drug development. [Read 1/36; Taft 1/7.23-7.25]
279. A compound can be cleared from the blood and excreted from the body unchanged. Clearance (Cl) is a critical determinant of what happens to a drug in the body, because it is the parameter (along with bioavailability) that determines the systemic exposure (AUC) following oral dosing, as described by the following equation: [Read 1/37; Taft 1/7.26(b)]
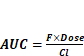
280. Clearance is defined as the volume of plasma from which the compound is completely removed per unit time, and is measured in mL/minute. However, clearance can be a slow process, and so the body uses metabolism to accelerate this. Metabolism is the enzymatic modification of compounds to increase clearance and occurs predominantly in the liver . In general, metabolism increases the polarity of a molecule by, for example, adding hydroxyls, conjugation with glucuronic acid, oxidising, and removing alkyl groups. The more polar products usually then have higher aqueous solubility, making them more readily excreted via the urine or bile. Predicting where on a compound metabolism will occur can be difficult, although certain groups (e.g. phenols) are known to be more likely to be metabolised. In 2001, there were standard and routine in vitro assays used to provide indications of metabolic stability. These included microsomal and hepatocyte stability assays where the compound is incubated with liver enzymes or cells, and then it is determined how much of the original compound remains after a set time period. [Read 1/37; Taft 1/7.26(b)]
281. Drugs with higher Cl will have lower systemic exposure (AUC) for a given dose, meaning lower drug levels in the plasma after a drug is administered. Compounds with high clearance from the body may therefore not remain in the blood long enough to effectively reach their target. [Taft 1/7.26(b)]
282. The clearance of many drugs is restricted by plasma binding and is directly proportional to the fraction of unbound drug in the plasma. These drugs are referred to as “low extraction ratio” drugs. Extraction ratio is the ratio of a drug’s metabolic Cl and hepatic blood flow (90 L/hr in humans). This ratio ranges between 0 (no metabolism or very low Cl) up to 1 (when Cl is so high it becomes limited by liver blood flow). [Taft 1/7.26(b)]
283. Hepatic metabolism can limit oral bioavailability through the “first pass effect”. Thus, drugs with a large metabolic Cl will have low oral bioavailability, because the fraction of the dose absorbed into the bloodstream that escapes first pass metabolism (fh) is low. This is illustrated by the following equation: [Taft 1/7.26(b)]
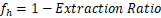
CNS Penetration
284. For targets not in the CNS, it is preferable to minimise the amount of compound reaching the brain. At the early stages of drug discovery, compounds could be evaluated via their physicochemical properties (e.g. polar surface area) to assess the likelihood of passing through the blood-brain barrier. At a later stage in development, there would be in vivo brain/blood ratio experiments in mouse or rat to determine brain penetration. These experiments would be run by (or at the direction of) the skilled DMPK scientist. [Read 1/39]
Drug-drug Interactions
285. Many patients will need to take more than one drug at the same time, particularly if they take a drug long-term for treatment or prevention of a chronic condition. Therefore, it is important to avoid drug-drug (and drug-food) interactions. Drug-drug interactions typically occur when one drug blocks or upregulates the enzymes that metabolise the second drug, making the half-life of the second drug unpredictable, which could result in loss of efficacy, or toxicity. One of the major concerns for drug-drug interactions is the inhibition or induction of cytochrome P450 (“CYP 450”), one of the major metabolising enzyme families in the liver. Therefore, it is important to minimise inhibition or induction of the major isoforms[11] of this enzyme, and CYP3A4 in particular. In 2001, in vitro assays were available to evaluate inhibition or induction of CYP 450 enzymes (and CYP3A4 in particular), although inhibition was far easier and cheaper to assess than induction and assessed earlier in the optimisation process. These assays would be done by (or at the direction of) the skilled DMPK scientist. [Read 1/40]
Chemical and metabolic stability
286. It is important to assess whether any of a potential drug’s metabolites or chemical degradation products are toxic. In particular, some compounds can form reactive metabolites where the metabolite is able to form a covalent bond with other proteins and DNA within the body. This process is referred to as bioactivation, and can lead to increased drug-drug interaction risk, and adverse side effects such as mutagenicity and idiosyncratic drug reactions. In 2001, in vitro assays were available to evaluate the potential for these liabilities. [Read 1/41]
Approach to Drug Development
287. Before a compound under development can be tested in humans, safety and proof of concept must be established through preclinical studies. Preclinical drug development involves using a combination of methods to assess physicochemical, pharmacologic, pharmacokinetic and toxicologic properties in order to screen out for further evaluation compounds with the optimal balance of potency, selectivity, safety and pharmacokinetics. [Taft 1/7.28]
288. The skilled DMPK scientist would work with the skilled pharmacologist and skilled medicinal chemist to determine when particular assays should be carried out to measure the DMPK properties of the compounds under investigation. Which particular assays would be used and in what order would depend on the target and what is discovered in earlier assays, with the priority and nature of the assays adapted dependent on the areas of concern that the previous screening of compounds had identified. [Read 1/42]
289. The skilled DMPK scientist would work with the skilled medicinal chemist to consider the results of any DMPK assays, and assess whether the compounds screened have properties which mean that further development of them is unlikely to be successful (e.g. high metabolic instability when incubated with hepatocytes) and/or could be modified to improve the DMPK properties of the candidate molecules (e.g. blocking a particular site on a molecule which is known to be liable to being metabolised (a so-called metabolic ‘hot spot’) while retaining potency against the target). [Read 1/43]
290. The amount of material needed to perform an in vivo preclinical pharmacokinetic experiment depends on the goal of the experiment and the species of animal tested. An initial screening experiment would likely involve administration of a low dose requiring a small amount of test material, this type of experiment would enable researchers to evaluate a number of compounds and to identify promising candidates that merit further pharmacokinetic evaluation. These most promising compounds would proceed to in vivo testing in animals. Taft 1/8.16]
291. Drug discovery and development is all about recognising the trade-offs between e.g. potency and other properties such as DMPK properties, and finding the correct balance between all the different properties. [Read 1/44; Taft 1/7.28]
