Remote hand-down: This judgment will be handed down remotely by circulation to the parties or their representatives by email and release to The National Archives.
Mr Justice Meade:
Introduction. 4
The issues. 5
The witnesses. 7
ACD's expert, Dr Wolf. 7
MI's expert, Prof Tyagi 8
The fact witnesses. 12
The skilled person. 12
The common general knowledge. 15
Law.. 15
Agreed CGK.. 16
CGK About Nucleic Acids. 16
CGK About Assays. 17
Hybridization assays. 19
Assays applied to extracted nucleic acid in vitro. 19
In situ hybridization. 20
Fluorescence in situ hybridization. 21
Probes. 22
Labels. 23
Multiplexing. 24
Amplification. 25
Blocking. 26
bDNA technical explanation. 27
Disputed CGK.. 29
The mindset point 29
bDNA.. 33
Cruciform probes. 35
The EP572 specification. 35
Claims in issue. 43
Infringement 45
The facts. 46
Evidence of what the skilled person would expect from the Patents. 52
The experiments. 53
Overview.. 54
The in situ experiments - assessment 57
Analysis. 61
Claim scope issues. 62
L sections complementary to non-overlapping regions. 62
Integer D(i) and G(i) of EP572, C(i) of EP439, "label probe". 66
Does the label probe have to be detectable on its own?. 67
Does the label probe have to be a terminating molecule?. 68
Analysis. 68
Integer D(ii) and G(ii) of EP572, C(ii) of EP439 ("amplifier") 69
Joint liability. 71
Relevant law.. 71
Facts as to MI's involvement with customers' use. 72
Validity. 74
Anticipation - the law.. 74
Obviousness - the law.. 75
Disclosure of Collins. 75
Disclosure of Kern. 79
Novelty analysis. 81
Obviousness over Collins and Kern. 83
Disclosure of Player 86
Obviousness over Player 86
Secondary evidence. 86
Insufficiency. 88
The Opposition Division Decision. 89
Conclusions. 89
1. In this action, the Claimant ("ACD") alleges that the Defendant ("MI") has infringed the following pair of European Patents (collectively "the Patents"):
i) European Patent (UK) No. 1 910 572 B1 ("EP572")
ii) European Patent (UK) No. 2 500 439 B1 ("EP439").
2. MI counterclaims for revocation of both Patents.
3. The Patents are from the same family and have the same, unchallenged, priority date of 20 June 2005 (the "Priority Date"). Their specifications are materially identical and the trial proceeded by reference to the specification of EP572; paragraph numbers in this judgment are to that specification. I set out the claims below but for present purposes just mention that EP572 has method claims and EP439 has product claims. This distinction makes no difference to the main validity attack, which is an obviousness one (with a sufficiency squeeze), but has potential implications for novelty and infringement, and for an added matter squeeze. The Patents concern in situ detection of nucleic acids by hybridisation. Below, I use "in situ" as an adjective and "ISH" as an acronym for "in situ hybridisation", and this is how the expressions were used at trial and in the literature. They are to some extent interchangeable, at least when the context implies the use of nucleic acid hybridisation, and thus "an in situ technique" and "an ISH technique" could be used to indicate the same thing.
4. Both parties are based in the US. ACD is a significantly larger company than MI. MI says that the UK proceedings are a "proxy" battle and that ACD has been forced to sue for infringement here because it has not (at least yet) been able to get any US patents granted, including on appeal at the PTAB. None of this is of any direct relevance to what I have to decide, but I will have to deal below with a point made by MI that a potential witness to ACD's case of secondary indicia of non-obviousness, a Dr Urdea, has been engaged to help ACD in the US but not called as a witness in this action.
5. MI denies infringement on the basis that its products and the methods for using them do not fall within the claims of the Patents. It also says that even if they do, it is not liable because it has not done and does not do any infringing act in the UK. It says that the products in question are sold in the US with title passing there, and are imported into the UK by its customers, not by it; and that its customers, not it, implement any method in the UK. ACD's response to this has been broadly twofold: first it says that title does not pass in the US so sales are made by MI in the UK, and secondly it says that MI is a joint tortfeasor with its (MI's) UK customers in relation to importation and use. The first of those would involve consideration of US law and other matters, and the parties agreed that it should be removed from this trial and determined at a later hearing if and as necessary and appropriate. The second, joint tortfeasorship, is within the scope of this trial.
6. The question of whether MI's products fall within the claims includes issues both of fact and of claim interpretation (with equivalence also in issue). In relation to the factual issues, MI relied on certain experiments. These were not litigation experiments but experiments done for product design and development in the course of MI's business. They occupied a significant but reasonable and proportionate amount of time at trial.
7. At trial, Mr Tom Moody-Stuart KC represented ACD and undertook the great majority of the oral advocacy; he led Mr Stuart Baran, who dealt with the added matter squeeze in closing oral submissions. Mr Thomas Hinchliffe KC undertook all the oral advocacy for MI, leading Mr Thomas Lunt. I note that Mr Moody-Stuart mentioned that ACD had had regard to the encouragement in the Patents Court Guide for parties to make greater use of junior advocates in deciding how to arrange its representation. I am grateful for this but make it clear that it was understandable and appropriate that MI did not take the same course; the added matter squeeze was tightly integrated with other issues and it was an equally valid choice to have the same advocate deal with all the issues. As it so happens, the added matter squeeze fell away entirely.
8. Each side called one expert witness. MI called three fact witnesses, covering the issues over relationships with its customers, and the experiments.
9. I am grateful to both parties and all their representatives for the way in which their cases were presented. The Agreed Statement of Common General Knowledge ("ASCGK") was very useful and also concise, given the high technical complexity, and the identification of the disputed CGK issues was precise and helpful. I found the materials on the experiments initially rather hard to follow and to marshal and at my request the parties cooperated during trial to produce some agreed summaries and diagrams which met this difficulty admirably.
10. Formally speaking, the issues are:
i) The identity of the skilled person.
ii) The scope of the CGK. Taken with the issue over the skilled person the main point on CGK is of high importance to the obviousness arguments.
iii) Four points of claim construction, which go essentially to infringement.
iv) A factual dispute about whether the alleged infringing products possess a particular advantage which, to the extent made out, MI says goes to infringement.
v) Whether the matters complained of fall within the Patents' claims:
a) On a "normal" construction;
b) By equivalence.
vi) Whether, if the claims do cover the acts complained of, MI is liable as a joint tortfeasor with its customers. This involves some limited fact-finding, although there is little if any dispute about it and the real argument is how properly to characterise, as a matter of law, what has been done.
vii) Anticipation by:
a) Collins et al, "A branched DNA signal amplification assay for quantification of nucleic acid targets below 100 molecules/ml", Nucleic Acid Rec. 1997; 25(15): 2979-2984 ("Collins");
b) Collins in combination with Kern et al, "An Enhanced-Sensitivity Branched-DNA Assay for Quantification of Human Immunodeficiency Virus Type 1 RNA in Plasma" J Clin Microbiol 1996; 34(12): 3196-3202 ("Kern");
viii) Obviousness over:
a) Collins;
b) Collins with Kern;
c) US Patent No. 5 635 352 ("Urdea");
d) Player et al, "Single-copy Gene Detection Using Branched DNA (bDNA) In Situ Hybridisation" J Histochem Cytochem 2001; 49(5):603-612 ("Player").
ix) Insufficiency under two heads, both squeezes with the obviousness attacks. A further free-standing allegation was dropped by MI in closing.
x) An added matter squeeze against infringement.
And in relation to obviousness and anticipation ACD relied on a decision of the Opposition Division of the European Patent Office upholding EP439.
11. Although all those issues remained live at the start of oral closing submissions in a formal sense, certain of them then fell away:
i) For reasons addressed below, ACD accepts that Collins and Kern may be read together for obviousness but not (at least to the extent necessary) for anticipation. So I have to deal with Collins on its own and in combination with Kern on anticipation, but for obviousness I need only, in practical terms, to consider them together.
ii) Urdea was maintained by MI as a back-up in the event that ACD were to argue that two features (cruciform probes and bDNA - see below) were not at least obvious to combine from Collins. ACD did not so argue and on that basis Urdea adds nothing and I need not rule on it.
iii) Obviousness over Player depends entirely on the state of the CGK (whether cruciform probes were CGK).
iv) The added matter squeeze was run by MI only in the event that ACD took a particular argument on infringement (that there was an amplifier that was capable of hybridising to one and only one label probe). ACD did not run any such argument so the squeeze was not needed and I do not have to decide it.
12. Each side called one expert. ACD's expert was Dr Catherine Wolf and MI's expert was Prof Sanjay Tyagi.
13. As fact witnesses, MI called:
i) Dr Harry Choi;
ii) Prof Niles Pierce; and
iii) Ms Gillian Pierce.
14. Dr Wolf is currently, and has for many years been, an independent consultant working on diagnostic methods.
15. In the course of that work, she consulted for ACD through an entity called Halteres Associates, with which Dr Urdea was also closely involved.
16. Prior to her consultancy work, Dr Wolf worked at Ventana Medical Systems. It seems that her roles there involved both business and technical functions.
17. Dr Wolf's academic background (at least in her postgraduate work) was in molecular and cell biology and focused significantly on ISH assays.
18. Dr Wolf gave her evidence in English (both orally and in her reports). That is not her mother tongue and at her request an interpreter was available, not to translate all the questions and answers but to step in in the event of problems. I am very grateful to the interpreter, but her assistance was called for on only a tiny number of occasions, and I find that Dr Wolf was entirely able to understand the questions in full and give her answers without any loss of understanding. It was faintly suggested by ACD that allowance should be made for her having additional stress caused by the language arrangements. I did not observe that and reject it.
19. MI made no personal criticism of Dr Wolf, but made the following submissions about her experience and background:
i) First, that she was not an expert in probe design. I agree with this to some extent, but the detailed design of actual probes at the nucleotide level is not important to the issues I have to decide, and at the level at which the Patents and the prior art are expressed I find that she understood matters amply well enough to help me.
ii) Second, that she gained experience of ISH in the years up to 1993, and thereafter was in business rather than technical roles such that her attitudes remained those of 1993, over a decade before the Priority Date. I reject this. The balance between business and technical matters in her work is not easy to define with precision but it was clear that she kept abreast of technical matters easily enough for her understanding of ISH to remain current.
iii) Third, that she was a "glass half empty" person. I reject any suggestion that she was, personally, of a pessimistic point of view, but I do agree that she tended to assess whether an assay approach would work by reference to very challenging goals, such as detection or quantification of very low levels of nucleic acids. The Patents' claims are not limited to such difficult tasks, and nor are such tasks the only goal of the prior art.
iv) Fourth, that she gave some long answers. However, MI also said that it was not submitting that the long answers were anything other than her trying to explain her position in her own way, so there is nothing to this. For what it is worth, I did not think her answers were all that long, other than one particular instance where she was simply, and helpfully, trying to pull together all the problems she saw with the ISH experiments put forward by MI, and I agree that Dr Wolf was trying to explain her position in her own way.
v) Fifth, that she said in her oral evidence that she had not had a chance to explain her position fully. She had had an adequate opportunity across her three reports and in oral evidence, but nothing turns on this and in any case I think that what she primarily had in mind was that she had not drawn together all her points on the experiments, which she addressed in the very long answer to which I have referred above.
vi) Sixth, that she had a habit of adding "possibly" or the like to the end of answers. It was submitted for MI that this had the effect of watering down concessions that she made that a particular technique would be expected to work. Instead of saying that it would, she was saying that it would, "possibly". I agree that she said this sort of thing quite often, to MI's frustration, but that is because she genuinely did not agree that things were so straightforward as MI's case required.
20. Overall therefore, I find nothing in those six points to lead me to downgrade Dr Wolf's evidence in general. She was adequately qualified to give her opinions and she did so honestly and directly. There is something in the third point - that she had in mind unusually difficult applications of the techniques in question - and I will take that into account in the specific context where it matters, which is primarily the question of reasonable prospects of success in relation to obviousness.
21. Following a distinguished career, Prof Tyagi is currently a Professor of Medicine in the New Jersey Medical School of Rutgers University.
22. Prof Tyagi's work has not been entirely academic, however. He has acted as a consultant to industry on a number of occasions.
23. Prof Tyagi has been involved in the development of new probe technologies. At the Priority Date this concerned in vitro development. At that time he had experience of in situ techniques, but this was experience of using them in his own research, not developing them.
24. ACD submitted that Prof Tyagi came to ISH development only ten years or so after the Priority Date, and that he was accordingly not in tune with the practical issues that faced such work at the Priority Date. Instead, ACD submitted, he had to try to put himself in the position as it was at the Priority Date by reading the literature, and that he had an overly expansive attitude to what was CGK.
25. I agree that Prof Tyagi was not specifically working on ISH probe development until after the Priority Date, but on the other hand he was working on in vitro assay development and was a user of ISH techniques. I also do not think that ACD put its finger on anything transformational in the art that took place in the period following the Priority Date and before Prof Tyagi's ISH development work started, at least not such as to make his task of reading in an unduly challenging one. I think the task of reading in was more a question of contextualising and cross-checking expertise and knowledge from work closely related in time and content, and I think he did a more than adequate job of it.
26. While making no personal criticism of him, ACD made a major attack on Prof Tyagi's evidence on the basis of hindsight in the light of the way he was instructed and the sequence in which he saw materials. There were two main limbs to this:
i) That he saw the Player prior art first, and therefore was liable to have carried what it showed (that bDNA could be used in situ) over into his assessment of the other, in vitro, prior art;
ii) That he was aware of the technology developed both by ACD and by MI before his involvement in the case.
27. ACD also relied on the way in which Prof Tyagi conducted literature searches of his own.
28. There have been many decisions about "sequential unmasking" in the instruction of experts in patent cases. I need not go into them any more than to say that it is an ideal for an expert giving evidence as to the obviousness of a patent to set out their views on the CGK and then assess what the prior art discloses and renders obvious, and only then to see the patents in suit. Parties instructing such experts are well advised to bring this about when possible.
29. However, sometimes it is not possible. Frequent reasons are that the invention of the patent in question is well known to be used in a commercial product, or that the expert has worked with one or other of the parties, or has been instructed in other proceedings. When it is not possible the Court has to take into account that there was a risk of hindsight - but the risk may not have eventuated.
30. Such is the case here: Prof Tyagi did indeed know of the parties' technologies. He therefore did not come to prepare his evidence in ignorance of the possibility of the in situ use of cruciform probes with amplifiers such as bDNA. I therefore have to ask myself, and consider carefully, whether his evidence on obviousness suffered from hindsight.
31. I did not detect any sign of hindsight, however.
32. But in any event, in the context of this case particularly (and unusually) the effect of hindsight, had it occurred, would have been much less material than usual. This may sound odd, so I will explain it a little further.
33. In the present case, the main obviousness case is over Collins with Kern. It is accepted by ACD that they would lead the skilled person to consider the possibility (although not, ACD says, to the standard of disclosure for anticipation) of using the bDNA assay disclosed with cruciform probes in situ. The only question is whether the skilled person, once they knew that idea, would consider that it had reasonable prospects of success. If ACD's defence to obviousness had been that cruciform probes and in situ use of bDNA were each individually known but that it required insight to have the idea of combining them, then hindsight could be much more important.
34. Similarly, it is true that Prof Tyagi saw Player first and that that shows the use of bDNA in situ, but for the same reasons it is not so very important because it is accepted by ACD that the idea of using bDNA in situ would occur to the reader of Collins and Kern. I would also observe that advisers to parties attacking a patent have to show the prior art to a potential expert one piece at a time and it is inevitable that such an expert will have seen prior art A before seeing prior art B. Sequential unmasking is important where it can be achieved but a judge should not expect the impossible. Had MI's advisers shown Prof Tyagi Kern before Player then no doubt ACD would have criticised that too.
35. This analysis would reduce the importance which hindsight would have had if I had thought there was any, but would not eliminate it. It could always be a material part of the background and ACD sought to deploy it in two additional ways:
i) First, that Prof Tyagi addressed the question of reasonable expectation of success on the basis that he knew that bDNA with cruciform probes in situ was in fact possible. This is true but in context of no real weight given my findings that Player was CGK and/or that the skilled person would have found it on a literature search. Even aside from those points I would not have attached much importance to it in the absence of some point being raised as to why bDNA would positively not be expected to work in situ.
ii) Second, that Prof Tyagi was affected by hindsight in his identification of the CGK.
36. As to the second of those points - hindsight to identify CGK - I think there are two matters to bear in mind:
i) It cannot have realistically involved hindsight for Prof Tyagi at least to ask himself the question of whether the matters discussed in Kern and Collins (cruciform probes, bDNA, use in situ) were CGK since the documents specifically mention them. It might be otherwise if he had spontaneously said that some feature missing from the prior art and which formed the bridge to the invention was CGK so as to make a conclusion of obviousness, but that is not this case.
ii) When it comes to assessing whether or not something was obvious over the prior art, and the reasons that an expert gives, the Court is inquiring into the thought processes of the expert. But when the Court is asking whether something was CGK or not, it is usually conducting a review of much more objectively ascertainable matters: what was in the literature and how widely it was circulated.
37. ACD also pointed to the fact that MI's solicitors provided some materials relevant to what might be CGK to Prof Tyagi, including after he had seen the prior art. Relatedly, it pointed out that they sent Prof Tyagi such materials on cruciform probes after he had seen Kern and therefore (ACD submitted) after he had decided, based on Kern, that they were CGK. I think this was an unreasonable point; the patent litigation process involves the expert identifying what in his or her opinion was CGK and this must of course be done fairly, but after the expert's views have been obtained then, anticipating that the other side will challenge them, historical materials demonstrating whether the opinion is right or wrong have to be gathered. This is usually a cooperative process, with the expert providing some materials and guidance about what would be CGK sources, and the advisers asking questions and sometimes, as in this case, obtaining materials which are hard to get for some reason or another. It is just not reasonable or practicable for an expert to get all the materials going to validate whether something is CGK before seeing the prior art.
38. Prof Tyagi was also criticised in relation to an article by Nolte. The article was not CGK, and MI did not argue that it was, it just deployed the article as part of its "weight of numbers" CGK argument on cruciform probes. I do not think that Prof Tyagi's views were based on the premise that it was itself CGK. Its contents were consistent with what he said was CGK, but there were adequate other materials supportive of his views on each point. I do accept that Prof Tyagi did not fully explain how he found Nolte, and he was mistaken in saying that he found it from its being cited in Urdea. It is possible that he found it from the Patents, but that was not demonstrated. Had the premise of Nolte itself being CGK been a material part of Prof Tyagi's views, or had it been more central to his overall opinions, this might all have been more important, but although ACD devoted quite a bit of attention to it, I think it was a minor part of the picture.
39. Taking all these matters together I do not think there was any culpable or material hindsight infecting Prof Tyagi's views on the prior art or CGK. Of course that does not in itself mean those views are correct. He might yet be wrong as to whether something was CGK (and indeed I have held that although he thought cruciform probes were CGK, they were not) but I think he was asking himself the questions in a fair way, representative of the skilled person, and not just because he knew what the invention of the Patents was.
40. Separately, ACD said that Prof Tyagi had in mind that the notional skilled person was capable of at least a little bit of invention. This was based on a passage of cross examination at T3/469, but on a fair reading I am sure the Professor did not mean that. There is one isolated answer which reads that way, but other questions and responses show that it was not so.
41. In terms of his general demeanour, Prof Tyagi was a model witness. His answers were extremely concise and to the point and he expressed his reasoning with great clarity. I found his evidence highly reliable and of great assistance. It is no personal criticism of her, but I found Prof Tyagi considerably better in expressing his reasons than Dr Wolf and while far from conclusive it is a reason in favour of preferring his evidence.
42. No criticism was made of any of MI's fact witnesses and I found each of them honest and fair.
43. I remind myself that Prof Pierce and Dr Choi were witnesses of fact and not expert witnesses (although they undoubtedly possess sufficient expertise to be such).
44. ACD pointed out that the MI fact witnesses are not neutral or independent, and cannot be expected to be. This is true but of no relevance. There is minimal if any factual dispute about the matters to which they spoke and even if there were, their evidence was not in any material way unbalanced, or said to be so.
45. The parties agreed that the proper approach was to identify the problem which the Patents aim to solve, and then to consider what the "established field" which existed was in which the problem was actually located. This involves looking at the actual facts as they existed at the Priority Date, including, among other things, what real teams there were. See in Illumina v Latvia MGI [2021] EWHC 57 (Pat) as applied in Alcon v Actavis [2021] EWHC 1026 (Pat).
46. The parties' rival positions were:
i) ACD and Dr Wolf said that the skilled person was someone interested in developing and/or using an ISH assay suitable for localising and quantifying low levels of target nucleic acids;
ii) MI and Prof Tyagi said that the skilled person was someone with an interest in developing techniques for nucleic acid detection.
47. The position of ACD and Dr Wolf is clearly incorrect in its limitation to quantifying low levels of target (and low levels of target generally), since the Patents' claims contain no such limitation, since the claimed method and products could be used for other less demanding purposes, and since neither side suggested that there was any real world team with such a narrow focus. Localisation is more reasonable since that is an inherent objective of many ISH approaches. But in any event the debate about the standard to be aimed for was not the central bone of contention on the identity of the skilled person: that was whether or not the skilled person was an in situ person (ACD's position) or had the more general areas of interest (nucleic acid detection generally) contended for by MI. The standard to be aimed for (whether or not the skilled person was interested specifically in the more difficult goals) came back in in relation to obviousness and I return to that below.
48. As to the problem which the Patents aim to solve, I find that it is the provision of better specificity and sensitivity in in situ hybridisation assays. The claims relate to in situ assays, and the early parts of the specification are directed to that area (see [0001] and [0007] to [0010]), as well as other later paragraphs in the specification (e.g. [0036], [0199]-[0202]). Implementing the solution requires the use of labelling and/or amplifying (including optionally pre-amplifying) but that is a necessary practical adjunct and not revealing of the central problem. I return to that point when seeking to identify the inventive core for the purposes of addressing infringement by equivalence.
49. Although MI rightly took the point that the problem is not specific to low levels of target, it did agree that the problem related to in situ detection. It took its stand on the proposition that the established field in which that problem resided was that of nucleic acid detection generally.
50. Turning to the real world situation, the following matters were relied on by the parties:
i) MI said that there was no field of ISH in itself, no "wall" (Prof Tyagi's phrase) between ISH and other nucleic acid detection approaches. In my view this largely just restates the question.
ii) MI said that the fundamental underlying molecular biology is the same between ISH and in vitro approaches: hybridisation, melting temperature, the need for a label and the like. It also pointed out that the same general categories of label (enzyme, fluorophores etc) are used. This is all true but of relatively low weight as there is no reason why two fields with the same underlying fundamentals could not be separate.
iii) MI said that numerous in vitro techniques had been transferred to the in situ context, including:
a) The original ISH itself;
b) Rolling circle amplification;
c) Branched DNA;
d) In situ PCR;
e) Tyramide signal amplification;
f) Padlock probes; and
g) Molecular beacons.
ACD disputed the details of some of these, as to the success they had enjoyed or whether or to what extent they were CGK. I deal with some of those points below, but whatever their detailed disposition there can be no doubt that there was an established pattern of at least trying to take, and at least on occasions succeeding in taking, techniques from in vitro to in situ. I find that this is an important and telling point in MI's favour. Indeed, I find it inconsistent for ACD to argue that there was a positive prejudice in the ISH field against the feasibility of taking in vitro techniques across to in situ while saying that the skilled person was confined to ISH and hence did not have CGK about in vitro techniques. It seems that the prejudice would positively require knowledge of the in vitro techniques and the difficulties in transposing them to in situ use.
iv) MI argued that there were companies, institutions and individuals working across in situ and in vitro applications, including Dr Wolf, Ventana and Bayer (MI also relied on Prof Tyagi himself but in my view he was at the Priority Date only a user and not a developer of in situ techniques). ACD joined issue on some of the examples given and I find that it probably was the case that some companies, perhaps Gen-Probe, only produced commercial results in the in situ field, but ACD did not dispute that there were other companies that were active in both in vitro and in situ and that is another powerful point in MI's favour.
v) As well as disputing MI's contentions, ACD put forward points of its own. In particular it relied on texts, reviews and even (it said) books which covered only in situ assays. This has some force, but only a little. The texts did not shun in vitro work, they just did not cover it. It is going too far to say, as ACD does, that Prof Tyagi really accepted that they treated ISH as distinct. The fact that there might be whole books about capacitors would not in itself prevent a relevant field being electronics generally, if other factors favoured that conclusion.
51. Balancing these factors, I come down firmly in favour of MI on this issue. The skilled person would be one who was involved with nucleic acid detection more generally. That would cover, but not be limited to, ISH.
52. Although I agree with MI on the point, and although a deal of time at trial was spent on it, I do not in the end find it all that important to my overall conclusions. It was said by ACD, who mainly made a feature of it, that it went to the CGK and to whether Prof Tyagi could adequately put himself in the position of the skilled person. Apart from the mindset against transposing techniques from in vitro to in situ, CGK is not very important to the main obviousness attack because of the fact that Collins and Kern point to all the features of the claims of the Patents, and I have found that Prof Tyagi's experience and approach did not prevent him from an adequate consideration of issues arising with in situ assays. The other potentially key place that CGK comes in is as to whether cruciform probes were CGK, since if they were then the attack over Player would succeed (and if not, not), but I am against MI on that anyway, regardless of the decision on the skilled person.
53. I note that most of the cross-examination of Dr Wolf took place on the assumption of her vision of the skilled person, and that further reduces any practical importance of this point.
54. In keeping with current practice in the Patents Court, there was a joint document which showed the CGK that was agreed (the "ASCGK") and another identifying what was in dispute. There were a number of listed issues of CGK in dispute; I have considered them under three headings below: some from the parties' list have dropped away and I found it convenient to group others together. The ASCGK also contained useful explanations of technical matters on topics where there was no dispute about how the technology worked, but where there was a dispute about whether it was CGK, in particular bDNA and cruciform probes.
55. There was no general dispute about the law applicable to CGK: to be CGK something must be generally known and accepted as a good basis for future action.
56. In the present case, the idea of mindset is an important one, heavily relied on by ACD. It pointed to Rockwater v Technip [2004] EWCA Civ 381 at [10] for the proposition that the skilled person will "share the common prejudices or conservatism which prevail in the art concerned".
57. I accept this as a general proposition of law. It implies there can be what might be called negative common general knowledge (see Dyson v Hoover [2002] RPC 22 at [55]-[57] where the art was "bag-ridden") and it bridges into the point that invention can lie "in finding out that that which those in the art thought ought not to be done, ought to be done" (Jacob J, as he then was, in Union Carbide v BP Chemicals [2007] FSR 37 at [25]-[28]).
58. Later, Jacob LJ in Pozzoli v BDMO ([2007] EWCA Civ 588) explained how this made sense analytically:
27. Patentability is justified because the prior idea which was thought not to work must, as a piece of prior art, be taken as it would be understood by the person skilled in the art. He will read it with the prejudice of such a person. So that which forms part of the state of the art really consists of two things in combination, the idea and the prejudice that it would not work or be impractical. A patentee who contributes something new by showing that, contrary to the mistaken prejudice, the idea will work or is practical has shown something new. He has shown that an apparent "lion in the path" is merely a paper tiger. Then his contribution is novel and non-obvious and he deserves his patent.
59. So one has to inquire whether a prejudice is part of the state of the art, and for most purposes it will be so, if it is, because it is CGK.
60. The EPO recognises the concept of a prejudice, too: see The Case Law of the Boards of Appeal of the European Patent Office, 10th Ed, 2022, at section 10.2. It requires a relevant prejudice to be "a widely held but incorrect opinion of a technical fact"; "an opinion or preconceived idea widely or universally held by experts in the field". The EPO requires a high standard of proof of a prejudice, the burden lying on the patentee to show it. This all seems to me to be consistent with the UK position, although I did not hear argument about the EPO case law specifically and I do not consider it necessary or right to hold ACD to any greater than the normal civil standard of proof of the prejudice alleged in this case as a matter of CGK.
61. Counsel for ACD suggested that the question of prejudice is best considered as a facet of the skilled person. For myself I think it is an aspect (a negative one, if proved) of the CGK, but it does not make any practical difference since the CGK is an integral part of the skilled person as a hypothetical construct.
62. MI relied on the fact that it can be CGK that a technique has been proposed as a way forward, even without it having in fact been demonstrated to work, on the basis that it is part of the skilled person's mental equipment not that that technique does work, but on the basis that it may (see Conor v Angiotech [2007] RPC 20 at [18]). I accept the existence of the principle but it does not disturb the broader notion that CGK must be generally known and generally accepted (albeit in this more nuanced sense) and it should not be allowed to make every attempted technique CGK. This mainly goes to the use of bDNA in situ.
63. DNA is a large, naturally occurring polymeric molecule which stores genetic information. The monomers which form DNA are called nucleotides. Each nucleotide consists of a nitrogenous heterocyclic base (or nucleobase). There are four naturally occurring nucleobases: adenine, cytosine, guanine and thymine (which are abbreviated to A, C, G and T, respectively). Nucleotides can link to each other to form very long strings of nucleic acids. Typically, two DNA strands naturally form a double stranded helix. This occurs by the nucleobases interacting, via hydrogen bonds, with corresponding nucleobases on the other strand of DNA, and thereby forming the double-stranded DNA molecule.
64. In DNA, adenine (A) only pairs with thymine (T) and cytosine (C) only pairs with guanine (G). The nucleotides which contain these nucleobases are said to be 'complementary' to each other.
65. When double-stranded DNA is heated above a characteristic temperature, this structure collapses such that the two complementary strands of DNA separate.
66. Ribonucleic acid (RNA) is another type of polymer also formed of nucleic acids. However, the nucleobases may be adenine, cytosine, guanine (in common with DNA) or uracil (which is the unmethylated form of thymine).
67. RNA is usually a single-stranded molecule unlike double-stranded DNA.
68. There are three major types of RNA which are termed 'ribosomal RNA' (rRNA), 'transfer RNA' (tRNA) and 'messenger RNA' (mRNA). They are each involved in protein synthesis:
i) mRNA is created by the process of transcription in which a section of DNA (a gene) is converted into mRNA by an enzyme.
ii) The mRNA produced by this process resembles the DNA sense strand and carries the information needed for protein synthesis in its base sequence.
iii) In turn, mRNA directs the synthesis of proteins, in the process known as translation. A group of three nucleotides on mRNA form a 'codon'.
iv) The codon binds specifically to a molecule of tRNA which presents the complementary 'anticodon'. That molecule of tRNA will carry the amino acid which corresponds to the codon on the mRNA. In this way, the identity of the amino acid which is presented by the tRNA is dictated by the codon on the mRNA. As the mRNA passes through the ribosome, the ribosome transfers the tRNA's appended amino acid to the growing polypeptide chain.
69. In addition to the four nucleobases described above, tRNA comprises many modified bases. Isocytidine (isoC) and isoguanosine (isoG) are examples of nonnatural nucelotides. It was known that isoC and isoG do not base-pair with natural nucleotides but do base-pair with each other (like e.g. how C pairs with G).
70. Whether DNA or RNA, polynucleic acids may be referred to as polynucleotides since they are polymers formed by the monomeric nucleotides.
71. Certain terms used in work concerning nucleic acid detection and which the skilled person would have been aware of are:
Accuracy – an assay is accurate when it yields a result close to a true or accepted value.
Background signal – background signal (or non-specific signal) is detected when molecular detection probes bind to anything other than the intended target nucleic acid sequence and may be generated both from non-specific binding and non-specific hybridization. Reducing nonspecific signal generated in an assay is a key consideration in nucleic acid detection, regardless of its source.
Detection limit – the detection limit (or limit of detection or limit of estimated detection) is considered as the lowest target amount or concentration of target that can reliably or consistently be detected.
Melting temperature - the two strands of a hybrid can be separated by increasing the temperature. The melting temperature refers to the temperature at which half of the hybrid molecules in a given sample are separated.
Non-specific binding – non-specific binding occurs when probes bind to sites other than the target molecule. The nature of the non-specific sites is often not known, but can be any component of the sample whether nucleic acid or otherwise, for example, probes binding to solid surfaces.
Non-specific hybridization – non-specific hybridization occurs when probes bind to strands of nucleic acid other than the targets intended by the assay.
Oligonucleotide - polynucleotides composed of relatively few nucleotides may be called oligonucleotides.
Precision – an assay is precise if multiple repeat experiments produce results close to each other.
Sensitivity – the more sensitive an assay is, the lower the amount of target which can be detected. The limit of detection is the lowest number of target molecules which can be reliably detected.
Signal or target signal – the signal or target signal represents the indication of target nucleic acid accurately being detected by a particular assay.
Signal to background ratio – the signal to background ratio is a parameter used to define assay performance. It can be increased by increasing the target signal, by decreasing background signal or by increasing the target signal and decreasing the background signal together.
Specificity – an assay is specific to a particular target if that target is detected preferentially over alternative possible nucleic acid sequences (not intended to be targeted). The number of false positives in an assay with high specificity is lower than an assay which is less specific.
Sensitivity and specificity
72. The desire of assay developers to increase sensitivity and specificity was a core driver to the development of assays. As the technology evolved, users wanted to be able to detect target sequences of interest when there were fewer and fewer copies of the target present, in particular RNA / DNA samples (i.e. by using assays with high sensitivity). For example, this was to ensure that low levels of pathogens could be detected, as pathogens present even in low levels can cause disease. They also wanted to ensure that non-specific hybridization and non-specific binding were minimised (i.e. by using assays with a high specificity) given that false detection of non-target nucleic acid sequences (or other non-target sites) would create false positives and lead to amplification of background signal rather than target signal. It was also helpful to users for tests to be relatively simple so that they could be run on multiple samples with relative ease. Improving the specificity and/or sensitivity of an assay should be the goal of any good molecular scientist.
73. To improve sensitivity it is necessary to increase the signal to background ratio when the number of target molecules is small, which can be achieved, for example, by improving specificity (i.e. by reducing background). In practice, reducing background was an ongoing challenge for assay designers before the Priority Date.
74. A hybridization assay is an assay used to detect particular strands of nucleic acids in a sample. In principle, the assay operates by the use of an oligonucleotide probe which is designed to be complementary to the target of interest that is presumed to be present within the sample. A nucleic acid sequence in the target of interest must therefore be known. If the presumed target is present within the sample, the probe should hybridize (i.e. bind) to it. The probe is or can be labelled so that a signal is detected, indicating the presence of the target. If the presumed target is not present within the sample, the probe is intended not to bind to any other components of the assay such that no signal would be detected.
75. The majority of in vitro hybridization techniques are applied to nucleic acids extracted from their source ("Extracted In Vitro" techniques). Extracted In Vitro techniques do not allow nucleic acids to be appreciated in their wider histological context, because they are not detected in situ, but after extraction from the sample, which involves complete disruption of the structure of the sample containing the target (sample 'grinding').
76. Extracted DNA fragments may then be separated by size using electrophoresis on a filter or membrane. When applied to DNA, this technique is known as Southern blotting. The same technique can be applied to isolated RNA extracted from samples (termed Northern blotting).
77. Alternatively, extracted nucleic samples can be placed and imaged in a slide well (referred to as 'solution-phase hybridization'). In these assays, the nucleic acids are captured onto a solid support, for example via capture probes which are oligonucleotide probes attached to the support at one end and designed to hybridize to the target at a separate portion of the capture probe. Labelled probes are introduced into the sample, and will hybridize to the target if it is present among the captured nucleic acids. The microwells are then washed with buffer. If the presumed target is present, the hybridized labelled probes should be retained and thereby allow a signal to be detected; if not, the labelled probes that remain unhybridized will be washed away and no signal detected. The washing step is therefore important because it removes any unbound label that would otherwise be detected (and which would therefore give rise to background signal).
78. A typical application of a solid support hybridization assay by the Priority Date was for use in determining whether a patient is infected with a transmissible viral disease such as HIV. In HIV-infected patients, virions and viral cells will be present within their blood. These viral particles carry the genetic information of the virus - i.e. the HIV genome. To determine whether a patient is infected, a blood sample is drawn and the cells are collected by a low speed centrifugation process and then lysed by addition of a lysis agent such as guanidine thiocyanate. The lysis agent breaks down the cell membranes and brings all nucleic acids (including the HIV genome) into solution. Once the target nucleic acid is bound to the solid support, labelled probes which are complementary to a portion of the HIV genome are added. If the patient has a sufficient quantity of HIV-infected cells, the result will indicate that the patient is infected and this assay may be used to quantify the number of HIV genomes detected.
79. Another format is a hybridization assay that is performed in situ.
80. In situ hybridization (or 'ISH') seeks to preserve the architecture of the biological sample with the aim of providing information about the presence, quantity and spatial arrangement of the target polynucleotide within the sample. The report of an ISH assay was first published in 1969. ISH was well-established at the Priority Date.
81. In ISH, nucleic acid expression can be visualised while the integrity of the cell (i.e. the cell structure) and/or tissue sample is preserved. ISH was commonly conducted using a tissue slice, a whole embryo or in cells. This means that the target nucleic acid can be identified and located within a cell, ensuring results can be seen in their morphological context.
82. The tissue sample is 'fixed' using chemical agents such as formaldehyde or glutaraldehyde which causes proteins within the sample to cross-link through their amino acids, thereby stabilising the morphology of the sample. Cells within the tissue slice are then 'permeabilized' using other agents such as alcohol or a detergent which degrade the cell membranes and thereby permit the labelled probes to access any target polynucleotides at their original locations within the tissue sample.
83. Following addition of the probes and incubation to allow hybridization, the tissue sample is washed using buffer. Washing steps are added to this process to remove substances that might disrupt the final ISH image. Washing steps may remove cell artefacts and excess probes that remain unbound after the hybridization step. It is important to avoid disrupting cell morphology (which may be caused by an excessive number of washes). Permeabilization and washing steps would have been well known to the skilled person and routinely adopted by the Priority Date.
84. ISH can be performed in whole embryos of organisms, such as fruit flies. These embryos are often so small that preparing a tissue slide is a practical challenge. In that case, the embryos are not mounted on a glass slide but suspended in solution during the hybridization. They too are fixed and permeabilised before probes are introduced and the washing steps are performed.
85. Alternatively, cells in samples of serum, urine, blood and pleural fluid can be gathered together using cytocentrifugation.
86. Fluorescence in situ hybridization ("FISH") is a form of ISH using fluorescent labels to generate signal and was commonly used at the Priority Date for RNA detection. It was developed in the early 1980's as an alternative to methods which used radiolabelled probes. Apart from the use of signal probes carrying different labels, the assay is performed in generally the same way as described above in relation to ISH.
87. The assay format - whether using a solid support or in situ – is typically determined by the nature of the application, particularly the composition of the biological sample and the information which the user wishes to determine. Seeking to determine simply the presence of an infection may be achieved by detecting viral nucleic acid within the blood using a solid support assay, whereas analysis of solid tumor tissue, for example, would lend itself better to an in situ assay performed on the tissue slice.
88. While the conditions for hybridization applied directly to RNA or DNA were well characterised at the Priority Date, hybridization in cell preparations and/or tissue samples require a user to review and balance a range of variables.
89. These variables include:
i) The effect of tissue or cell preparation on the retention and accessibility of target nucleic acids;
ii) The effect of the hybridization conditions on the efficiency of hybridization in the context of the need to retain the structural integrity of the sample; and
iii) The construction of the probe itself, which impacts the efficiency of probe labelling, penetration and sensitivity of the method used for signal detection.
90. The impact of these variables on Extracted In Vitro assays differs from that on ISH assays:
91. Samples are prepared for Extracted In Vitro assays by 'grinding' or otherwise disrupting the sample so that nucleic acid can be extracted from the cell. Unlike ISH assays, cell structure and morphology need not be maintained, so the sample can be completely broken down: making the nucleic acid accessible, and not 'contained' within the sample.
92. Hybridization efficiency can be sped up in Extracted In Vitro assays by agitation of a sample, a condition that may be applied easily to techniques applied to extracted nucleic acids, as samples presented in a well or on a filter can be shaken to increase the speed of hybridization. Agitation is not suitable for some ISH samples.
93. Probe construction must be considered: see below.
94. Probes can be defined according to the type of nucleic acid they are made of and the method by which they are made. Each probe type has different characteristics. For example, RNA probes can form very stable hybrids, but can be difficult to prepare and store due to their inherent instability; in contrast, they can carry several label molecules. Oligonucleotide probes (single-stranded synthesised DNA with a sequence complementary to the target nucleic acid) are very stable, and can be produced in large amounts. However, due to the short length of these probes, they can carry only few labels.
95. One can have DNA–DNA, DNA–RNA, RNA–DNA, and RNA–RNA ISH, depending upon the probes and targets concerned. The likelihood of a target nucleic acid and probe nucleic acid sequence annealing and separating depends upon various factors, including temperature, the nature of the probes and target molecules, and the composition of the hybridization (salt and formamide concentration) and washing solutions.
96. The melting temperature depends on various factors.
i) Usually, sodium chloride is used as the ion provider, and an increase in sodium concentration will facilitate hybridization, while a decrease will lead to separation of the strands, influencing the 'stringency' applied in the hybridization step.
ii) %GC, which is the percentage of guanosine and cytosine in the probe molecule.
iii) The longer the probe, the higher the melting temperature of the hybrid.
iv) Percentage of formamide in the hybridization solution.
v) Percentage of non-complementary bases between the probe and the target molecule. The greater the mismatch between probe and target nucleic acid, the lower the melting temperature and strength of the hybrid.
97. Probe length impacts the 'strength' of nucleic acid/ target hybrids. Increasing probe length has other implications for ISH assays, which must be balanced against the benefits that might be conferred by increasing hybrid 'strength'.
98. In the direct labelling technique, the size of the label molecule used will restrict how many labels can be incorporated along the length of the probe. Longer probes can therefore contain more labels: the longer the probe, the greater the potential signal it may generate per probe. In relation to specificity, whilst it is correct that a probe of 20 nucleotides will likely be more specific than a probe of 10 nucleotides (because within a sample there may be multiple examples, other than the target nucleic acid, of the same sequences of 10 nucleic acid bases), generally a probe of 200 nucleotides in length will be less specific than a probe of 20 nucleotides because the tendency for the probe to bind to non-target sequences is increased with the longer probe. Ultimately, the decision as to what length of probe the assay developer would seek to develop will depend on the chosen assay conditions and other factors such as the number of labels to be incorporated. At the Priority Date the general tendency was for in situ hybridization assay designers to use small probes, typically of less than 100 nucleotides in length. These probe lengths (of generally less than 100 nucleotides in length) are generally chosen to optimise specificity (although probes greater than 100 nucleotides had been successfully used).
99. High temperatures are not necessarily damaging to cell structures. It was known at the Priority Date that as part of the sample preparation steps, fixed tissue samples could be subjected to temperatures of up to 95 degrees Celsius for a short amount of time without causing damage.
100. Extraction of nucleic acid from a sample means that excess proteins can be removed, so the final processed sample should predominantly consist of the selected type of nucleic acids (RNA is destroyed prior to DNA analysis; DNA is destroyed prior to RNA analysis). This reduces the number of molecules that may interact with longer probes to produce background signal. In contrast, ISH assays retain the proteins and cellular components available to interact with long probes and create background noise. Longer probes are also more likely to bind to non-target nucleic acids within the cell; forming a sufficient number of hydrogen bonds that the off-target hybrids persist to the visualisation stage. This creates a greater background signal that must be distinguished from on-target binding. The density of background-generating molecules are lower in Extracted In Vitro assays as opposed to ISH assays, due to the way these samples are prepared.
101. Further, the nucleic acid sequence used by the probe must have a low degree of homology with other, non-target, sequences to minimise off-target binding and ensure specificity. The number of 'unique' sequences available to image a target is dependent on the target sequence itself.
102. In high stringency conditions, which include high temperatures and a low concentration of salt in the hybridization buffer, only highly similar nucleic acid sequences bind. When the conditions of stringency are too high, any probe will separate from its target nucleic acid. Low hybridization temperatures and high concentrations of salt in buffers create low stringency conditions. These conditions enable the hybridization of less homologous nucleic acid sequences. However, when the conditions of stringency are too low, non-specific hybridization occurs and probes bind off-target.
103. Ultimately the choice of probe length will be designed whilst having the hybridization conditions in mind. For example, by varying the temperature and salt concentration, in situ hybridization assays can be optimised as appropriate.
104. Detection of nucleic acid targets could be achieved using a number of different labels. Sometimes the labels were enzymes, such as horse radish peroxidase, which convert a colorless substrate to a colored product, or a non-fluorescent substrate to a fluorescent product, or enzymes such as alkaline phosphatase that catalyzes a reaction that generates light (chemiluminescence). The labels were designed to create a signal which could be recorded for example using a microscope equipped with a digital camera or a luminometer.
105. Early labels were radioactive nucleotides that contained a radioactive isotope. Those labels were detected using appropriate sensors however, owing to their inherent risk (and other reasons) they were replaced with other labels, for example fluorescent labels or enzyme labels.
106. A fluorescent label incorporates a fluorophore which, when excited by light of a particular colour, emits light in a second colour, which is detected.
107. In situ hybridization assays typically use chromogenic or fluorescence labels and use microscopes which provide spatial information about the location of the target in the context of the tissue. However, in situations where cells are present in suspension, e.g. during analysis of blood cells, in situ hybridization assays may use flow cytometers where a cell's total fluorescence is recorded as it moves past a recorder.
108. Probes can be detected through direct or indirect methods. In the direct approach, nucleotides are modified to contain a label molecule (e.g. an isotope for radioactive probes, or a fluorophore for fluorescent probes). These modified nucleotides are incorporated along the length, or at the tail, of the label probe. As the labelled molecule is bound directly to the target and directly produces a signal, the resulting hybrid can be detected immediately after post-hybridization washes. In contrast, indirect labels, such as biotin or digoxygenin, are not fluorescent or radioactive, but are then detected through a reaction, or a chain reaction with other molecules that will create the ultimate signal (fluorescence, for instance).
109. Enzymes are usually indirectly linked to the probe and can be visualised through the application of a substrate that results in a chromogenic reaction.
110. Fluorescent labels, unlike enzyme labels, give a signal without the need to apply further reactants.
111. Fluorescent labels provided users of ISH technology the ability to 'multiplex': imaging multiple targets within the same cell or tissue sample (using distinguishable labels e.g. fluorophores which emit different coloured light). Previously, only one radioisotope might be used on a sample at a time. To identify three different targets, three different slides would have to be used, all of which would potentially contain different cells and/or different parts of cells.
112. At the Priority Date, many researchers wished to carry out multiplexing assays to compare the expressions of many genes in the same sample and obtain gene expression profiles of different cell types to characterize them.
113. A limitation of multiplex detection was the inability of digital microscopes to reliably distinguish between more than 4 or 5 colours. Approaches to overcome this disadvantage included labelling a probe with more than one fluorophore and associating a colour combination with a particular target.
114. Multiplex detection would typically be expected to give rise to greater levels of background, since the greater number and types of probes could interact with each other and bind or hybridize non-specifically to other components within the assay. In situ assays inherently have a greater degree of background owing to the extent of cellular (non-target) tissue.
115. The skilled person would have been aware of the desire for multiplexing assays at the Priority Date, and the corresponding need to ensure that assays were specific and sensitive. Multiplexing was a procedure that was well known at the Priority Date.
116. Polymerase chain reaction ('PCR') was introduced in 1985 as a method to amplify target strands of DNA and created possibilities of molecular diagnostics for pathogens. In PCR, a thermostable DNA polymerase enzyme is used in a combination with a pair of primers to synthesize iteratively two strands of a portion of the DNA. The DNA sample is first thermally denatured by heating it above its melting temperature such that the two strands of DNA are separated from each other. Each of the primers anneal to the separated strands of DNA. The DNA polymerase will locate the ends of the short double stranded regions of DNA where the primers have bound and then move along the DNA, adding the correct complementary nucleotide to the original DNA strand. The result of this is the creation of a double stranded DNA, which is a replica of the original double stranded DNA used in the experiment. By iterating this process multiple times, the region of DNA bounded by the two primers is amplified exponentially (1 DNA sample becomes 2 in the first PCR process, 2 become 4 in the second, 4 become 8 in the third etc.). Although originally PCR required the addition of fresh DNA polymerase during each cycle (because the thermal denaturing of the DNA also denatured the DNA polymerase), at the Priority Date thermally stable DNA polymerases (such as Taq, the DNA polymerase from the bacterium Thermus aquaticus) were commonly used, enabling automation of the process. The development of PCR as an amplification technique was considered a key milestone in the development of amplification methods, in Extracted In Vitro assays. PCR utilises polymerase enzymes to create more nucleic acid and was used to increase the amount of target nucleic acid extracted from tissues or cells.
117. A development to PCR technology included amplification from RNA (as opposed to DNA).
118. PCR allowed the detection of pathogens by rapidly identifying their DNA/RNA sequences within a few hours, thereby reducing the inherent risks of contamination associated with traditional culturing methods. However there were limitations:
i) First, to discern the amplified product, gel electrophoresis followed by hybridization was required. This process resulted in the dispersion of amplified DNA within the laboratory, creating a significant potential for carryover contamination in subsequent assays;
ii) Second, thermal cycling was required, which could be expensive in comparison to isothermal amplification methods; and
iii) Third, targets often contained single nucleotide polymorphisms (i.e. mutations) and if they occurred in the region where primers (and/or probes in the case of real-time PCR, as described in the following paragraph) bind, no signal would be created.
119. Real-time PCR was developed to solve the limitation of having to do electrophoresis followed by hybridization and associated dispersion of DNA. In real-time PCR fluorescent probes that detect and indicate the amplified DNA in sealed tubes are used and the detection is carried out in the tubes as the PCR progresses. This format avoids contamination issues and increases the dynamic range of the assays.
120. In order to obtain spatial information from tissue sections in situ PCR was also developed. This technique is performed on a section and amplified DNA is detected. However, this technology had limitations because amplified DNA would diffuse away from the target site and the tissue architecture was degraded due to thermal cycling. Ultimately, by the Priority Date there was not significant interest in the field in in situ PCR.
121. One of the main approaches used at the Priority Date was multilayer detection, in which a marker is applied to the probe, for example digoxigenin, and then an antibody is added on top of that. Further antibodies are added, building an enlarged compound before a final layer producing the detectable signal (enzymatic or fluorescent), is added.
122. Tyramide signal amplification is another example of what might be used. The probe label is detected through a combination of multi-layered antibodies, and a final powerful enzymatic reaction triggers a high amount of coloured or fluorescent compound from an initial tyramine complex.
123. Rolling circle amplification was also employed, which utilised a circular template of single stranded nucleic acid, consisting of a sequence that binds to a target sequence, a primer and a sequence that will bind to a label probe. The template will first bind to the target nucleic acid sequence. A primer then binds to the circular template, enabling the start of a 'chain reaction' initiated by a polymerase enzyme, which moves along the template and synthesises a single stranded nucleic acid consisting multiple repeats of the template sequence upon which further (and further) label probes may bind. This increases the signal generated from the assay. Unlike PCR amplification, which needs to undergo cycles of heating and cooling, rolling circle amplification has the advantage of being isothermal, therefore better preserving the morphology of the sample (the central purpose of the ISH assay).
124. In the context of a hybridization assay, binding of probes to non-specific sites (i.e. those other than the target sites) can be suppressed to some extent using "blockers". For example, blockers can be proteins which occupy the non-specific sites but do not themselves bind to the specific target sites of interest, or salmon sperm DNA that is easily extractable and has a low degree of homology with human DNA. In this way they decrease the propensity of non-specific sites giving rise to background signal.
125. Some commonly used blockers were dried milk powder or purified agents such as bovine serum albumen (BSA).
126. The following is an explanation of bDNA. It is a matter of dispute whether bDNA, especially in situ, was CGK. But, as I have mentioned above, the way in which it works is common ground, so I explain it here (and the same applies to cruciform probes, also covered in the following).
127. In bDNA amplification, signals are amplified rather than the target or probes. The technique involves hybridizing target nucleic acids with multiple sets of branched DNA probes that form tree-like structures that can carry a large number of signaling moieties. Heavily labelled probes would either be bound directly to the target or were bound to the target via secondary probes called preamplifiers.
128. In a bDNA assay multiple target specific probes are generally used to capture the target nucleic acid (DNA or RNA) onto the surface of a microtiter well plate. A second set of target probes (label extenders) hybridize to the target site and in the first generation assays, also serve as binding sites for the amplifier molecules. The branched DNA probe features arms, each of which carries alkaline phosphatase reporter molecules (i.e. the labels). This approach, which creates something akin to a Christmas tree with ornaments, facilitates significant signal amplification at each target probe site and allows a large number of enzyme-labelled probes to be hybridized to each target molecule in this manner.
129. A representation of a bDNA assay is depicted in, among others, Nolte (the status of Nolte in the CGK debate was also in dispute, but its depiction is useful):
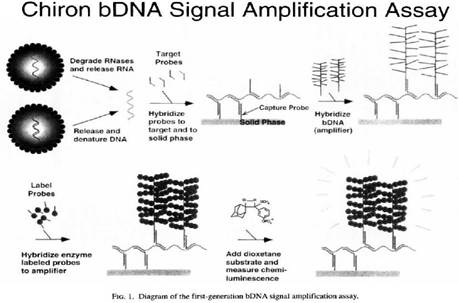
Figure 3
130. It can be seen from Figure 3 that the target DNA is part of a sandwich format in which the target (1) indirectly hybridizes to capture probes which are attached to (and stick out from) the 'Solid Phase' surface; and (2) indirectly hybridizes to the branched DNA amplifier. Enzyme labelled probes are hybridized to the arms of the branched DNA amplifier, before the addition of dioxetane, which is a substrate for the enzyme (alkaline phosphatase), and a measurement of light (chemiluminescence) created during the enzymatic reaction is taken.
131. The bDNA assay was developed in the second and third generation assays to increase the number of labelled probes that could be bound to the target. This was achieved through the addition of preamplifier molecules which would hybridize (1) indirectly to the target (via the label extender); and (2) to the amplifier molecule carrying the enzyme labelled probes. The skilled person would be aware that this would allow lower quantities of target RNA or DNA to be detected i.e. provide an improved sensitivity, although as signal strengths are increased by the use of probes with a larger number of labelled probes, the likelihood of generation of background signals (false positives) would also increase due to their binding in a non-specific manner.
132. The so-called "cruciform" design is also something that it is necessary to understand for the purposes of this case, and as with bDNA generally it is disputed whether it was CGK, but how it works is not.
133. A cruciform design using two label extenders was also introduced in the second generation of bDNA assay, to reduce background by eliminating signals generated from non-specific binding of single label extenders and these were incorporated into Chiron/Bayer's commercial products. A representation of a bDNA assay with the cruciform arrangement is depicted in, among others, Weikersheimer et al. (Exhibit ST-8):
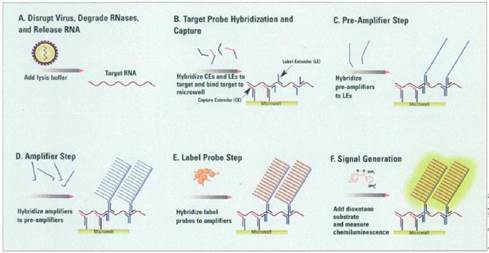
Figure 4
134. In the third generation of bDNA assay the non-natural bases isocytidine (isoC) and isoguanosine (isoG) were incorporated into the amplification probes. The use of isoC and isoG containing probes in the bDNA assays increased the target-specific amplification without a concomitant increase in the background from nontarget sequences.
135. By the Priority Date, a number of papers had demonstrated the use of bDNA in in situ assays (again, it is disputed whether this was CGK).
136. As I have said above, I identify three disputed areas which I deal with in turn. In places I have identified review and other articles and materials relied on by the parties. I have not attempted to cover all such materials raised at trial in this judgment, although I reviewed what was said in the written closings about them, but have focused on the ones which I found most helpful and/or which were relied on most strongly by the parties.
137. The alleged mindset is not about real difficulties that would be experienced by the skilled person in trying to implement the teaching of Collins and Kern as to bDNA with cruciform probes in situ. ACD does not assert the existence of any actual problem that would prevent success (and this is emphasised by the fact that Player, Kenny, Chiron and ACD, in its work subsequent to the Patents, all used straightforward conditions). Its case is that there was merely a perception of low/no expectation of success when seeking to take a technique from in vitro to in situ. Nor does ACD argue that the Patents actually provide a concrete solution to any particular problem of implementation, or a demonstration that the technique works in situ; the examples are prophetic. While ACD did not say that the conditions used in Player and/or Kenny (cited in the Patents) are unusual in themselves, it did argue that reference to them in the specification would dispel the low expectation of success represented by the mindset, simply by being a demonstration of the use of bDNA in situ (albeit without cruciform probes).
138. Three of the disputed CGK points in the parties' agreed list go together under this heading. They were numbers 1, 2 and 5 in the list:
1. The skilled person's CGK as to the similarities and differences between in situ hybridization and Extracted In Vitro techniques.
2. The skilled person's perception as to the extent to which detection and amplification principles, probe designs, amplification methods, reagents, and/or experimental conditions could be transferred from in vitro hybridization assays to in situ hybridization assays and the ease of doing so.
5. Whether the skilled person perceived that in situ hybridization assays were difficult to perform successfully because of the need to select suitable parameters for assay variables, including the skilled person's ability to optimise an in situ hybridization assay by adjusting those parameters.
139. In addressing these, I take into account the matters that I have considered in connection with the identity of the skilled person, above, in relation to techniques which had been transferred from the in vitro setting to the in situ setting.
140. As well as the identification of the three listed issues of disputed CGK, I find it helpful to set out what ACD says the mindset or prejudice was. In its closing written submissions it said this (at paragraph 52):
The skilled person would have approached the suggested transfer of an in vitro assay into ISH with significant caution and would not consider it had a reasonable prospect of success. Matters differ when in the case of an assay that has been shown to work in an ISH context.
141. And when I asked Counsel for ACD to encapsulate the mindset in a couple of sentences during oral closing arguments, he said this (T7/894):
MR. MOODY-STUART: The mindset is a scepticism that nucleic acid detection techniques that have previously been demonstrated in vitro, or indeed in any other field but in vitro here, could be transferred or could be used as the basis for a new in situ hybridisation technique without significant work and optimisation.
142. I would not think it fair to hold ACD to these statements as if they were a carefully drafted contract term or a statute, to be picked over for their fine details, but it seems to me that there is a substantive and important difference between them. The latter really just says that the skilled person would think that the transfer from in vitro to in situ would require significant work and optimisation, while the former says, actively, that there would not be an expectation of success. I find it hard to the point of impossible to see how the latter, even if it were shown to be the attitude of the art as a matter of CGK, could be a mindset that would deter the skilled person from going forward with development work in pursuit of taking an otherwise attractive proposition from in vitro to in situ. That is especially so in this art, where it is clear that getting hybridisation assays working well is fiddly, requires attention to numerous parameters and is very empirical. That does not make that sort of work inventive or a deterrent to trying to make progress; it is just the nature of the beast.
143. In any event, I will bear in mind both formulations of the mindset as proposed by ACD.
144. I also bear in mind the following general points:
i) To the extent there was a scepticism, it would quite logically have been all the greater when the skilled person was thinking about the task of getting to a really good, commercial ISH product, or to an ISH assay for more difficult tasks such as quantifying rare nucleic acids. That is not the right standard because the claims of the Patents are not so limited, but it was the standard that Dr Wolf was, at least often, applying.
ii) I am not just looking to identify the matters that would have to be dealt with to make an in situ assay work. I have to ask myself if the skilled person would have a pessimism about coping with them.
iii) I need to ask myself why there might be pessimism: might some facet of the task be seen as especially difficult for some reason, or was there a track record of failure on some particular matter?
iv) This is an inquiry into the CGK so I must have regard to what are the relevant CGK sources, especially, in the context of this case, as often, review articles and textbooks. I accept, however, ACD's submission that negative "Eeyorish" results and views are less likely to be reported.
145. I was referred to a number of review articles/book chapters on this issue, including the following:
i) Dirks, "RNA molecules lighting up under the microscope" Histochem Cell Biol (1996) 106:151-166.
ii) Polak, "In situ hybridisation: Principles and practice", 2nd Ed. (1998)
iii) Levsky and Singer, "Fluorescence in situ hybridisation: past, present and future", Journal of Cell Science 116, 2833-2838 (2003).
146. Dirks is of particular importance because ACD used it, helpfully, as a focus of its case. Dirks provides a useful list of the matters that would require attention in making an ISH assay work. There are seven of them and they are set out in the right hand column on the first page.
147. Similarly, Dr Wolf in her second report listed what she called "key determinants", at paragraph 4.18. MI summarised these, fairly in my view, as being:
(a) the probe size and ability to penetrate;
(b) ability of the signalling molecule to localise vs diffuse away;
(c) pre-treatment to fix and permeabilise, washing and assay temperature;
(d) hydrophobicity of non-natural nucleotides; and
(e) enzymatic reactions.
148. Item (d) has no relevance to the situation I have to consider because Dr Wolf accepted in cross-examination that it did not apply to isoC/isoG as used in Collins. Item (e) has no relevance because Dr Wolf said she had in mind potential problems with polymerase enzymes and not enzymatic labels such as alkaline phosphatase.
149. Counsel for MI went through (a) to (c) systematically in cross-examination by reference to a number of articles. Dr Wolf essentially accepted that there were routine ways to make choices for each of the parameters. For example, appropriate temperatures were to be found in Polak. I will not go through each of the points let alone all of the references because ACD's position in closing was not really to defend any of them individually.
150. Rather, in response, ACD submitted that Dr Wolf had not said those were the only factors, just the key ones. I agree that she made occasional statements that there might be other points, but it was her decision to put these forward as the key ones, and since MI challenged them all successfully, it is thoroughly unconvincing to say that I should take into account that there might possibly be others which would be more significant.
151. ACD also submitted that MI was "salami slicing", just dealing with the individual factors and ignoring the possibility that in aggregate the "key determinants" added up to justifying the mindset alleged. I certainly agree that I should see the picture in the round and I have aimed to do so. I also accept that in theory it might be possible for there to be a mindset that many small individual problems form an overall mass justifying pessimism, but that is not how Dr Wolf's evidence put it, and I find that ACD's salami-slicing point is an unconvincing ex-post reaction to the individual points' failing when tested in the oral evidence.
152. My reaction to Dirks, relied on particularly by ACD as I have said, is that it sets out in a careful and systematic way what the skilled person would need to consider and to do to make an ISH assay work (the focus is RNA specifically, but neither side said that that made a material difference). I think the statement in the Methodology section in the right hand column on the first page, following the list of 7 matters:
These steps have been extensively optimized to develop generally applicable protocols for sensitive and high resolution RNA-ISH on tissue sections and cell preparations.
fairly captures the attitude of the skilled person (there is a similar statement in the first sentence of the Conclusions section on the last page of the article). The message is that ISH works, that multiple factors have to be taken into account, that optimisation of them is needed, and that that has been done. It is by no means the message that the work is trivial, but there is no hint that a good result cannot be achieved by appropriate care and diligence. Despite being the high point or at least the focus of ACD's case it is nowhere near establishing a pessimistic mindset. I note that Dirks refers to the 7 matters as being interdependent and have taken that into account, but it does not detract from my overall view. ACD relied on the fact that the Dirks paper was "worth reporting". Had Dirks been a report of original experiments undertaking a new approach for the first time that could perhaps have had some weight, but it is a review paper, summarising the state of the field.
153. As well as this consideration of the key factors from Dr Wolf's evidence and from the literature, I think ACD's mindset argument is fatally undermined by the situations where techniques had been taken from in vitro to in situ with success. I have touched on these in dealing with the identity of the skilled person, above. ACD nibbled around the edges of these. For example:
i) It said that in situ PCR had "died a death" by the Priority Date. The position was that in situ PCR had been made to work, which contradicts ACD's mindset allegation, but had proved to have limitations (migration of DNA, issues with thermal cycling) that meant that it was less good than had been hoped, and was overtaken by other techniques. I agree that this might affect the skilled person's expectation that taking a new technique from in vitro to in situ would be a world-beater but I have rejected that as not being the right question. Anyway, the issues with in situ PCR were specific to the way that PCR works and could not have provided reasons for pessimism across the board.
ii) It said that rolling circle amplification had taken years to develop. That may be so, although the reasons for the timing were not well established in evidence, and I think it more likely that what took a long time was getting the technique to work to a very high standard.
154. But ACD's problem, it seemed to me, was that even if it undermined to some extent some of the instances where techniques had been transferred to the in situ situation, it could not hope to paint a picture that the skilled person would approach such a task without a reasonable expectation of success. The absolute maximum that it could hope to show was that the skilled person would not think that success was guaranteed. Even if that were so, it would not help ACD when it comes to concrete consideration of the obviousness argument over Collins/Kern: I have not approached that on the basis of the skilled person thinking that success was guaranteed.
155. On the basis of the above, I reject ACD's mindset contention.
156. The Patents themselves also undermine ACD's mindset argument. Player, Kenny and a US patent application (which is not relied on by ACD and was not the subject of any evidence) are referred to twice, at [0009] and at [0221], which I quote below. To run mindset successfully as an answer to obviousness, ACD needs to show that the Patents dispel the relevant prejudice, but these paragraphs do not indicate the existence of a prejudice or that it has been dispelled. Instead, they read as if the necessary conditions can be found by routine experiment or by consulting the literature in an ordinary way. A patentee is not bound by statements as to the CGK in a specification, and may in principle prove a different state of affairs, but it is a relevant factor and is against ACD in the present case. It is not, however, necessary to my decision and my finding above against the existence of the mindset alleged, which is founded on the expert evidence and the literature.
157. What bDNA is, is set out above. There was a dispute about whether bDNA in general was CGK and, more specifically, whether the use of bDNA in situ was CGK. The issue about bDNA generally is not of the greatest importance because all the cited prior art discloses it. The dispute about whether bDNA being used in situ was CGK is however important because although it is accepted to be an obvious thing for the skilled reader to contemplate starting from Collins and Kern, ACD then asserts the mindset point, and one way MI responds is by saying that successful use of bDNA in situ was CGK. I agree that that would be an effective response if there were a mindset (although I have rejected ACD's case on that) and ACD did not really argue otherwise.
158. MI's case on this point really cut to the chase, because its primary contention was that Player itself was CGK. MI's argument was based on the following:
i) Both experts read Player at the time.
ii) Player was picked up in review papers including Qian and Lloyd, "Recent Developments in Signal Amplification Methods for In Situ Hybridization", 2003 and Qian and Lloyd, "In Situ Hybridization: Basic Approaches and Recent Development", 2004. I find that these kinds of review paper are good evidence of the state of the CGK and that Dr Wolf accepted as much. Both these papers report that bDNA in situ had been used with success (not merely a paper proposal) and cite Player.
iii) Dr Wolf's evidence in cross-examination; MI submitted that she accepted that Player was CGK.
159. Points ii) and iii) both involve an assessment of short parts of the oral evidence of Dr Wolf (T2/184 and 259-260). I acknowledge that these are not completely unambiguous. Taken just as black and white words on the page the former is capable of being read merely as an acceptance that the Qian papers were good papers and the latter as being entirely premised on the Qian papers and being qualified or limited to Dr Wolf's own experience ("I would think so. I was made aware of the Player paper through Chiron ..."). I was well aware of the importance of this point from the parties' opening submissions and so paid close attention at the time to what Dr Wolf was saying, and I conclude without any real hesitation that she did mean to accept that Player was CGK. I am fortified in the conclusion that it was by the fact that I am not just relying on her say-so but on the objective support of the review papers. Some support is also to be had from the fact that Player is from Chiron/Bayer, a very significant company in the field.
160. In addition, I have Prof Tyagi's evidence to support my conclusion. He plainly was aware of bDNA as a general technique and I accept that he read Player at the time: it makes sense and there is nothing to gainsay it. I bear in mind that he had not used the technique himself and that is a factor, but no one in this field can possibly have used all the techniques that were in the CGK. It does not mean that there was no CGK. I also bear in mind that when he started considering what was CGK for his report he obtained Kern at an early stage, but I reject any suggestion that that in fact misled him about what the standard for CGK was or what was CGK. He did accept that Kenny and another paper called Antao would not be found without a literature search but it was not said that the same applied to Player.
161. The fact that Kenny and Antao would not be found without a literature search does not help ACD if Player was CGK. On a similar note, ACD pointed out that Prof Tyagi also identified review articles which do not mention the use of bDNA in situ, in particular a review by Tsongalis. That is nothing to the point.
162. I conclude that Player was CGK, and with it the fact that bDNA had successfully been used in situ.
163. This issue matters for the obviousness case over Player.
164. MI's evidence is much weaker here than on bDNA. There are various documents which refer to the use of cruciform probes in Chiron's commercial products but their quality is not comparable to, for example, the review papers that support MI's CGK argument on bDNA in situ.
165. I accept that sometimes CGK can be proved by showing a wide spread of references even if none individually is a typical CGK source. MI referred to my decision in Merck Sharp & Dohme v Wyeth [2020] EWHC 2636 (Pat) at [85]-[86]. I stand by what I said there, but it should be noted that I said that whether a spread of individual documents shows CGK depends on evidence about their having had "sufficient reach, impact and acceptance", and showing CGK from a variety of individual documents none of which is a usual kind of CGK source may well fall short. This point should not be allowed to water down the test for CGK so that anything that can be found by ex-post research in some (modest) number of documents is good enough.
166. The documents relied on by MI are relatively few and relate almost entirely to the Chiron product. Not all of them are even very clear about what the cruciform structure was and I accept ACD's point that a reader who did not already know what it was might well struggle to see or understand it. I also reject as unconvincing MI's reliance on post priority review papers. They may have heightened awareness at that later date but although they must have been written before the Priority Date, I do not think I can safely go further than to conclude that the individual authors knew about cruciform probes at the Priority Date.
167. MI's fallback was that if a skilled person were prompted to look into bDNA for any reason they would find out in due course about cruciform probes by looking through the literature. I think this is properly the province of an obviousness case over Player rather than a contention about CGK but in any event I found it vague and tenuous and I reject it.
168. I conclude that cruciform probes were not part of the CGK.
169. As I have said, the (undisputed) priority date is 20 June 2005.
170. Where I quote from the specification I have in places removed hyphenation from the original for readability and/or in light of the fact that the insertion of the text into this judgment has changed the line breaks. The same applies to some of the quotations from the cited prior art, below.
FIELD OF THE INVENTION
[0001] The invention relates generally to nucleic acid chemistry and biochemical assays. More particularly, the invention relates to methods for in situ detection of nucleic acid analytes in single cells. The invention also relates to detection and identification of single cells, particularly rare cells.
....
[0007] In situ hybridization (ISH) technology is an established method of localizing and detecting specific mRNA sequences in morphologically preserved tissue sections or cell preparations (Hicks et al., 2004). The most common specimens used are frozen sections, paraffin embedded sections or suspension cells that were cytospun onto glass slides and fixed with methanol. Detection is carried out using nucleic acid probes that are complementary to and hybridize with specific nucleotide sequences within cells and tissues. The sensitivity of the technique is such that threshold levels of detection are in the range of 10-20 copies of mRNA per cell.
[0008] However, ISH technology faces a number of technical challenges that limit its wide use. First of all, cells immobilized on solid surface exhibit poor hybridization kinetics. Secondly, assay optimization is generally required for a target mRNA in probe selection, labeling, and detection, for each tissue section in fixation and permeabilization, and in hybridization and washing. In addition, various experiments need to be performed to control for the specificity of the probe, for tissue mRNA quality, and for the hybridization efficacy of the experimental procedure. In addition to technical issues, current ISH technology has relatively low performance standards in term of its detection sensitivity and reproducibility. The false positive rate is still high unless the relevant cells are re-examined manually using their morphology, which is time and labor-intensive. Current ISH technology also does not have the capability to quantitatively determine the mRNA expression level or to simultaneously measure the expression of multiple target mRNA within cells, which may provide clinical valuable information such as increased detection sensitivity and specificity, and the identification of primary tumor type, source and stage.
[0009] There are four main types of probes that are typically used in performing in situ hybridization within cells: oligonucleotide probes (usually 20-40 bases in length), single-stranded DNA probes (200-500 bases in length), double stranded DNA probes, or RNA probes (200-5000 bases in length). RNA probes are currently the most widely used probes for in situ hybridization as they have the advantage that RNA-RNA hybrids are very thermostable and are resistant to digestion by RNases. RNA probe is a direct labeling method that suffers a number of difficulties. First, separate labeled probes have to be prepared for detecting each mRNA of interest. Second, it is technically difficult to detect the expression of multiple mRNA of interest in situ at the same time. As a result, only sequential detection of multiple mRNAs using different labeling methods has recently been reported (Schrock et al, 1996; Kosman et al, 2004). Furthermore, with direct labeling methods, there is no good way to control for potential cross-hybridization with non-specific sequences in cells. Branched DNA (bDNA) in situ hybridization is an indirect labeling method for detecting mRNA in single cells (Player et al, 2001; US 2002, 0172950). Branched DNA ISH has also been evaluated for detection of nucleic acids sequences in tissue specimens (Kenny et al, 2002). This method uses a series of oligonucleotide probes that have one portion hybridizing to the specific mRNA of interest and another portion hybridizing to the bDNA for signal amplification and detection. bDNA ISH has the advantage of using unlabeled oligonucleotide probes for detecting every mRNA of interest and the signal amplification and detection are generic components in the assay. However, the gene specific probes in the bDNA ISH need to be theoretically screened against possible non-specific hybridization interactions with other mRNA sequences in the cells. The nonspecific hybridization of the oligonucleotide probes in bDNA ISH can become a serious problem when multiple of those probes have to be used for the detection of low abundance mRNAs. Similarly, although use of bDNA ISH to detect or quantitate multiple mRNAs is desirable, such nonspecific hybridization of the oligonucleotide probes is a potential problem.
[0010] The present invention overcomes the above noted difficulties and provides methods for detecting nucleic acids in and for identifying individual cells. A complete understanding of the invention will be obtained upon review of the following.
....
[0030] In embodiments in which two or more first capture probes and/or two or more second capture probes are employed, the capture probes preferably hybridize to nonoverlapping polynucleotide sequences in their respective nucleic acid target.
171. There is a Definitions section starting at [0071]. The definitions relied on by the parties at trial are:
[0078] Two polynucleotides "hybridize" when they associate to form a stable duplex, e.g., under relevant assay conditions. Nucleic acids hybridize due to a variety of well characterized physico-chemical forces, such as hydrogen bonding, solvent exclusion, base stacking and the like. An extensive guide to the hybridization of nucleic acids is found in Tijssen (1993) Laboratory Techniques in Biochemistry and Molecular Biology- Hybridization with Nucleic Acid Probes, part I chapter 2, "Overview of principles of hybridization and the strategy of nucleic acid probe assays" (Elsevier, New York), as well as in Ausubel, infra.
...
[0081] The term "complementary" refers to a polynucleotide that forms a stable duplex with its "complement," e.g., under relevant assay conditions. Typically, two polynucleotide sequences that are complementary to each other have mismatches at less than about 20% of the bases, at less than about 10% of the bases, preferably at less than about 5% of the bases, and more preferably have no mismatches.
172. This is a particularly important definition for the parties' arguments. It is to be noted that "complementary" does not require 100% matches at the nucleotide level; indeed the definition is not couched in terms of the percentage match at all, but in terms of whether the polynucleotide will form a stable duplex with its complement. It is a functional, practical definition and not a numerical one.
173. The definitions relied on continue as follows:
[0083] The term "label probe" refers to an entity that binds to a target molecule, directly or indirectly, and enables the target to be detected, e.g., by a readout instrument. A label probe (or "LP") is typically a single-stranded polynucleotide that comprises at least one label which directly or indirectly provides a detectable signal. The label can be covalently attached to the polynucleotide, or the polynucleotide can be configured to bind to the label (e.g., a biotinylated polynucleotide can bind a streptavidin-associated label). The label probe can, for example, hybridize directly to a target nucleic acid, or it can hybridize to a nucleic acid that is in tum hybridized to the target nucleic acid or to one or more other nucleic acids that are hybridized to the nucleic acid. Thus, the label probe can comprise a polynucleotide sequence that is complementary to a polynucleotide sequence of the target nucleic acid, or it can comprise at least one poly-nucleotide sequence that is complementary to a polynucleotide sequence in a capture probe, amplifier, or the like.
[0085] An "amplifier" is a molecule, typically a polynucleotide, that is capable of hybridizing to multiple label probes. Typically, the amplifier hybridizes to multiple identical label probes. The amplifier also hybridizes to at least one capture probe or nucleic acid bound to a capture probe. For example, the amplifier can hybridize to at least one capture probe and to a plurality of label probes, or to a preamplifier and a plurality of label probes. The amplifier can be, e.g., a linear, forked, comb-like, or branched nucleic acid. As noted for all polynucleotides, the amplifier can include modified nucleotides and/or nonstandard internucleotide linkages as well as standard deoxyribonucleotides, ribonucleotides, and/or phosphodiester bonds. Suitable amplifiers are described, for example, in USPN 5,635,352, USPN 5,124,246, USPN 5,710,264, and USPN 5,849,481.
174. At paragraph [0102] there is a discussion of capture probes which was relied on by ACD and the subject of argument by both sides in relation to the "overlapping" claim construction points:
[0102] In methods in which two or more first capture probes and/or two or more second capture probes are employed, the capture probes preferably hybridize to nonoverlapping polynucleotide sequences in their respective nucleic acid target. The capture probes can, but need not, cover a contiguous region of the nucleic acid target. Blocking probes, polynucleotides which hybridize to regions of the nucleic acid target not occupied by capture probes, are optionally provided and hybridized to the target. For a given nucleic acid target, the corresponding capture probes and blocking probes are preferably complementary to physically distinct, nonoverlapping sequences in the nucleic acid target, which nonoverlapping sequences are preferably, but not necessarily, contiguous. Having the capture probes and optional blocking probes be contiguous with each other can in some embodiments enhance hybridization strength, remove secondary structure, and ensure more consistent and reproducible signal.
175. The points made concerned what would be conveyed by "contiguous", "distinct" and "non-overlapping" and in particular whether the latter two were being used merely tautologously, with both being deployed only for emphasis (MI's position), or if they meant different things (ACD's contention). I return to this point briefly when I deal with claim construction.
176. At [0104] the specification deals with the actual sequence of the capture probes, which was touched on in the written submissions but not a major focus of argument:
[0104] Potential capture probe sequences are optionally examined for possible interactions with non-corresponding nucleic acid targets, the preamplifiers, the amplifiers, the label probes, and/or any relevant genomic sequences, for example. Sequences expected to cross-hybridize with undesired nucleic acids are typically not selected for use in the capture probes. Examination can be, e.g., visual (e.g., visual examination for complementarity), computational (e.g., a BLAST search of the relevant genomic database, or computation and comparison of binding free energies), and/or experimental (e.g., cross- hybridization experiments). Label probe sequences are preferably similarly examined, to help minimize potential undesirable cross-hybridization.
177. There is some teaching about Fixation and Permeation at [0199]ff which was submitted by MI to show that the specification expects the skilled person to be able to find appropriate conditions using their CGK:
Fixation
[0199] In this step, the nucleic acids are immobilized within cells by cross-linking them within the cellular structure. There are a variety of well known methods to fix cells in suspension with a fixative reagent and to block the endogenous RNase activities, which can be adapted for use in the present invention. Fixative reagents include formalin (formaldehyde), paraformaldehyde, gluteraldehyde, ethanol, methanol, etc. One common fixative solution for tissue sections includes 0.25% gluteraldehyde and 4% paraformaldehyde in phosphate buffer. Another common fixative solution for tissue sections includes 50% ethanol, 10% formalin
[0200] (containing 37% formaldehyde), and 5% acetic acid. Different combinations of the fixative reagents at various concentrations are optionally tested to find the optimal composition for fixing cells in suspension, using techniques well known in the art. Duration of the fixing treatment can also be optimized. A number of different RNase inhibitors can be included in the fixative solution, such as RNAlater (Ambion), citric acid or LiCl, etc.
Permeation
[0201] Fixation results in cross-linking of the target nucleic acids with proteins or other cellular components within cells, which may hinder or prevent infiltration of the capture probes into the cells and mask the target molecules for hybridization. The assays of the invention thus typically include a follow-on permeation step to enable in-cell hybridization. One technique involves the application of heat for varying lengths of time to break the cross-linking. This has been demonstrated to increase the accessibility of the mRNA in the cells for hybridization. Detergents (e.g., Triton X-100 or SDS) and Proteinase K can also be used to increase the permeability of the fixed cells. Detergent treatment, usually with Triton X100 or SDS, is frequently used to permeate the membranes by extracting the lipids. Proteinase K is a nonspecific protease that is active over a wide pH range and is not easily inactivated. It is used to digest proteins that surround the target mRNA. Again, optimal concentrations and duration of treatment can be experimentally determined as is well known in the art. A cell washing step can follow, to remove the dissolved materials produced in the permeation step.
178. I accept the submission that the characteristics of the notional skilled person can be inferred to some extent from the tasks which the specification expects them to be able to do (see Horne v Reliance [2000] FSR 90) but it does not have much impact here because both sides said the skilled person would have aptitude in dealing with in situ experimental work.
179. In a section starting at [0204] the specification moves on to discuss probe design and selection by reference to certain of the figures. [0207] and Figure 4 are of particular importance to the construction issues ("LP" is label probe and "SGP" is signal-generating particle, abbreviations which were introduced in [0204]):
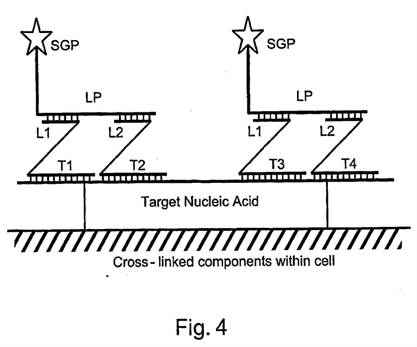
[0207] In a further indirect capture embodiment shown in Figure 4, two adjacent capture probes are incorporated in a probe set targeting a gene of interest. T1 and T2 are designed to be complementary to two unique and adjacent sections on the target nucleic acid L1 and L2, which can be different or the same, are complementary to two adjacent sections on the label probe. Their binding sections, T, L or both, are designed so that the linkage between the label probe and the target is unstable and tends to fall off at hybridization temperature when only one of the capture probes is in place. Such a design should enable exceptional specificity because the signal-generating label probe can only be attached to the target gene of interest when two independent capture probes both recognize the target and bind to the adjacent sequences or in very close proximity of the target gene. In a further embodiment, the melting temperature, Tm, of the T sections of the two capture probes are designed to be significantly above the hybridization temperature while the T m of the L sections is below the hybridization temperature. As a result, T sections bind to the target molecule strongly and stably during hybridization, while L sections bind to the label probe weakly and unstably if only one of the capture probes is present. However, if both capture probes are present, the combination of L1 and L2 holds the label probe strongly and stably during hybridization. In another embodiment, Tm of the T sections is below hybridization temperature while Tm of the L sections is substantially above. In the same way, the linkage between the label probe and the target can only survive the hybridization when both capture probes are hybridized to the target in a cooperative fashion.
180. It should be noted that this discussion covers both T1 and T2 where they hybridise to the target and L1 and L2 where they hybridise to the label probe. This needs to be borne in mind because "non-overlapping" is used to describe the regions of the target and of the label probe to which the capture probes hybridise.
181. In later figures, arrangements are shown in which amplification is used and there are accordingly many more SGPs, like leaves on a tree. For example, Figure 8 shows the following:
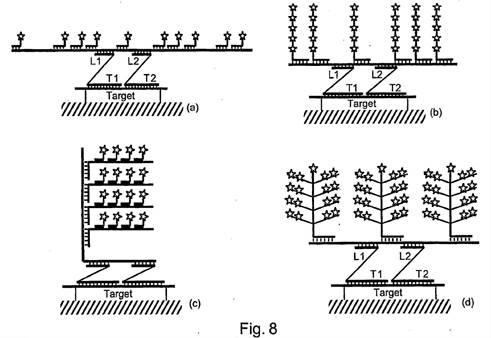
182. The specification gives information about hybridization conditions at [0221] and this is important to the arguments because it is the reference to Player and Kenny that ACD says breaks up the squeeze between obviousness and insufficiency:
Hybridization Conditions
[0221] The composition of the hybridization solution can affect efficiency of the hybridization process. Hybridization typically depends on the ability of the oligonucleotide to anneal to a complementary mRNA strand below its melting point (Tm). The value of the Tm is the temperature at which half of the oligonucleotide duplex is present in a single stranded form. The factors that influence the hybridization of the oligonucleotide probes to the target nucleic acids can include temperature, pH, monovalent cation concentration, presence of organic solvents, etc. A typical hybridization solution can contain some or all of the following reagents, e.g., dextran sulfate, formamide, DTT (dithiothreitol), SSC (NaCl plus sodium citrate), EDTA, etc. Other components can also be added to decrease the chance of nonspecific binding of the oligonucleotide probes, including, e.g., single-stranded DNA, tRNA acting as a carrier RNA, polyA, Denhardt's solution, etc. Exemplary hybridization conditions can be found in the art and/or determined empirically as well known in the art. See, e.g., U.S. patent application publication 2002/0172950, Player et al. (2001) J. Histochem. Cytochem. 49:603-611, and Kenny et al. (2002) J. Histochem. Cytochem. 50:1219-1227, which also describe fixation, permeabilization, and washing.
183. Claim 1 of EP572 is:
A. A method of detecting one or more nucleic acid targets within an individual cell, the method comprising:
B. providing a sample comprising the cell, which cell comprises or is suspected of comprising one or more nucleic acid targets;
C. fixing and permeabilizing the cell;
D. for each nucleic acid target, providing
(i) one or more label probes, wherein each label probe comprises one or more labels,
(ii) one or more label probes, and one or more amplifiers, wherein each label probe comprises one or more labels, and wherein each amplifier is capable of hybridizing to one or more label probes, or
(iii) one or more label probes, one or more amplifiers, and one or more preamplifiers, wherein each label probe comprises one or more labels, wherein each amplifier is capable of hybridizing to one or more label probes, and wherein each preamplifier is capable of hybridizing to one or more amplifiers;
E. for each nucleic acid target, providing two or more different capture probes,
i) wherein each of the two or more capture probes comprises a section T complementary to a section on the nucleic acid target and a section L complementary to a section on the label probe, or on an amplifier, or on a preamplifier, and
ii) wherein the T sections are complementary to nonoverlapping regions of the nucleic acid target, and the L sections are complementary to nonoverlapping regions of the label probe, the amplifier, or the preamplifier;
F. hybridizing, in the cell, the two or more capture probes to a single copy of the nucleic acid target, when present in the cell;
G. capturing the label probe to the two or more capture probes, thereby capturing the label probe to the nucleic acid target,
i) by simultaneously hybridizing at least two different capture probes to a single copy of the label probe,
ii) or by simultaneously hybridizing at least two different capture probes to a single copy of the amplifier and hybridizing the label probes to the amplifier,
iii) or by simultaneously hybridizing at least two different capture probes to a single copy of the preamplifier and hybridizing the one or more amplifiers to the preamplifier and the one or more label probes to each of the one or more amplifiers; and
H. detecting a signal from the label.
184. Claim 1 of EP439 is:
A. A kit for detecting a nucleic acid in an individual cell comprising, packaged in one or more containers:
B. at least one reagent for permeabilizing cells; at least one capture probe set comprising two or more capture probes capable of hybridizing to a target nucleic acid sequence; and
C.
(i) a label probe comprising a label, wherein the label probe is capable of hybridizing to said set of two or more capture probes, or
(ii) a label probe comprising a label, and an amplifier hybridized to the label probe and capable of hybridizing to said set of two or more capture probes, or
(iii) a label probe comprising a label, an amplifier hybridized to the label probe, and a preamplifier hybridized to the amplifier and capable of hybridizing to said set of two or more capture probes,
D. wherein each said capture probe comprises a T section which is complementary to a region of said target nucleic acid sequence and a L section which is complementary to a region of said (i) label probe, (ii) amplifier, or (iii) preamplifier;
E. wherein the T sections of the two or more capture probes in the capture probe set are complementary to the non-overlapping regions of the target nucleic acid sequence, and
F. the L sections of two or more capture probes in the capture probe set are complementary to the non-overlapping regions of said (i) label probe, (ii) amplifier, or (iii) preamplifier.
185. This is a product claim. It requires a kit packaged in (a) container(s) and that one of its components is a "permeabilising reagent". Those points may be relevant to anticipation and to infringement but there is no material difference between EP572 and EP439 when it comes to obviousness.
186. Formally speaking, claim 3 of EP439 was also in issue but I do not believe it adds anything and it was explicitly accepted not to add anything on obviousness, so cannot affect my overall conclusion.
187. There are many facets to the infringement case, and I think an overview will be helpful.
188. In terms of claim scope:
i) It will be seen from the claims of both patents that they each require one of three options to be chosen: (i) a label probe, (ii) a label probe plus an amplifier or (iii) a label probe plus an amplifier plus a preamplifier.
ii) ACD relies on options (i) and (ii) but not (iii).
iii) Whether these are present depends on points of claim interpretation about the meaning of "label probe" and "amplifier" as applied to one or more of the components of MI's products. But there is no dispute of fact.
iv) For option (i) and (ii), there is a requirement that the L sections of two or more capture probes are complementary to "non-overlapping" regions of the label probe or amplifier, as the case may be.
v) This is the central and overarching question of claim scope. It depends on claim interpretation and on equivalence. It also depends on a technical question about what the skilled person's expectation would be as to how a method which had an overlap would function, if it functioned at all.
189. For each of the disputed points of claim scope, their application to the facts depends on a detailed and careful understanding of how MI's system works. Nearly all of that is agreed, but there is one significant factual dispute: MI says that its system involves an "overlap" that makes a difference to the operation of the system. MI says that it makes the system better because it increases the specificity. ACD disputes that this advantage exists (as well as saying that even if it does, it does not matter). The advantage is referred to as the "Alleged Improvement" in some of the papers, and below.
190. To the extent that ACD succeeds on claim scope, it then has to show an infringing act. As explained in the introduction to this judgment, this trial is confined to the allegation that ACD is jointly liable with its customers for importation and use. I need to decide certain facts - basically how closely involved MI is with its customers' use of the MI system - and I then need to apply the legal principles, (as to which any dispute is minor at most), to them.
191. It is a principle that the claims of a patent should have the same meaning independently of the alleged infringement. At the same time, it is recognised that regard may need to be had to the alleged infringement so as to frame the relevant question (see Terrell at 9-65ff); this is such a case. I am therefore going to deal with the facts first. I caution myself that although it may be appropriate to proceed in this way to frame the relevant question, I should not, when it comes to it, make my decision on claim interpretation as if the skilled person had knowledge of the alleged infringement when seeking to understand the claims. The situation with equivalence is different, however, since the Actavis v Lilly questions require knowledge of the infringement.
192. MI produced a detailed PPD. Many of the details are not relevant. I will reproduce the most relevant diagrams and either provide my own narrative, or take the text from the PPD where greater technical detail and/or precision is needed.
193. The relevant products are called HCR 3.0 Products. They support the use of an RNA FISH assay (an in situ assay) according to the steps I will describe. A variety of Probe Sets may be used and I will describe different probes and the experiments that have been done on them below.
194. An HCR Probe Set has probe pairs p1 and p2. These can be seen in Figure 1 of the PPD:
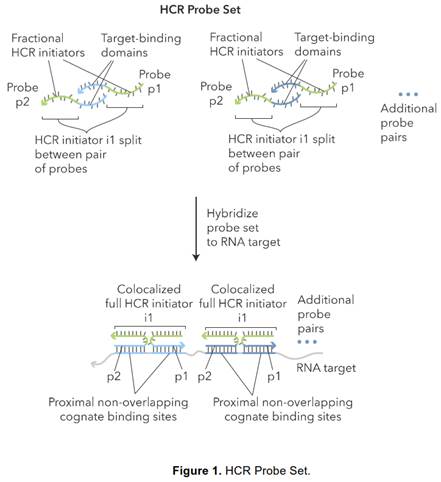
195. In the lower part of Figure 1 the probes p1 and p2 have bound to the RNA target. Their sequences are chosen so as to cause their binding close together on the target. There is no dispute that p1 and p2 are complementary to non-overlapping regions on the target; the infringement issue is about what happens next.
196. At this stage there is no label and no amplification has taken place. Again, that comes in succeeding steps, starting with the use of two HCR hairpins, h1 and h2. These are shown in figure 2 of the PPD:
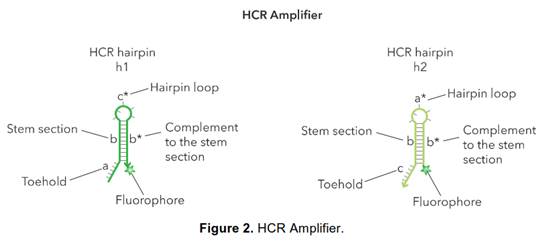
197. As will become apparent, the "Toehold" on h1 will hybridise to one end of the initiator that was to be seen in Figure 1 (the "Colocalized full HCR initiator", formed by the cooperation of the upper surfaces of p1 and p2), and then the hairpin h1 will unzip itself, with more and more of its length hybridising to the initiator. When it is unzipped, its base pairs will no longer be hybridised to one another internally to the hairpin, and will be available to hybridise to hairpin h2. A concatenation of h1 and h2 hairpins will form in a chain reaction, each hairpin with a label, and the labels will be used for detection.
198. In somewhat high level overview the components and a forming concatenation of h1s and h2s can be seen in Figure 3:
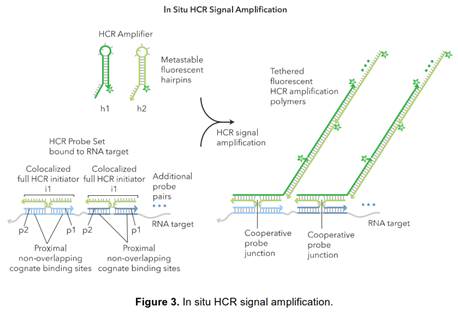
199. There is no dispute that this is a process of amplification, as the label to Figure 3 says. There is amplification because the initial attachment of one h1 leads to a string of multiple h1s and h2s, each with a fluorescent label.
200. The reason for the hairpin arrangement of the h1s and h2s (rather than having them just as linear single-stranded polynucleotides) is to prevent them hybridising to one another (or anything else) prior to the attachment of the probe pairs.
201. The facet of the MI system that gives rise to the infringement issue concerning "nonoverlapping regions" concerns the details of what happens when the unzipping of the hairpin encounters the place where the two probes p1 and p2 meet. This can be seen in Figure 5b of the PPD:
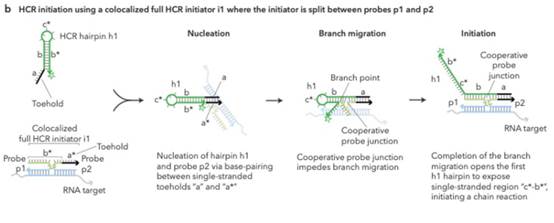
202. And as is explained in the PPD in the following terms:
"Using a colocalized full HCR initiator i1(Figure 5b), hairpin h1 nucleates with the initiator via base-pairing to single-stranded toehold "a" as before. A branch migration then begins in which the branch point again moves randomly a portion of the way along the stem as hairpin:hairpin base pairs are replaced by initiator:hairpin base pairs and vice versa. However, once the branch point reaches the junction between the two fractional initiators, there is an energetic barrier to proceeding further along the stem because formation of the next probe:hairpin base pair leads to formation of the cooperative probe junction that is energetically less favorable than the stacked hairpin:hairpin base pair it is replacing, somewhat impeding HCR initiation. As a result, weaker HCR signal amplification is achieved using probe pairs that colocalize a full HCR initiator i1 compared to using an HCR initiator i1 that is a single strand."
203. To put it in a very colloquial way, the unzipping has a tendency to "get stuck" when it encounters the "divot" where probe p1 and probe p2 meet. If this prevents the full unzipping and therefore the subsequent amplification then signal will be lost for that particular probe pair and performance degraded. It is essentially a matter of random chance whether this happens for any particular probe pair bound to the target so this effect leads to a reduction in signal in the system as a whole, not a complete loss.
204. This is where the "overlapping" issue comes in. MI found out (it seems by chance) that the energetic barrier that tended to cause the unzipping to "get stuck" could be ameliorated by having regions in the sequences of the probes p1 and p2 in which the nucleotides were complementary (in the sense of matching complementary nucleotides) to overlapping regions of the HCR hairpin h1. This can be seen, although not very easily I have to say, in Figure 6 of the PPD:
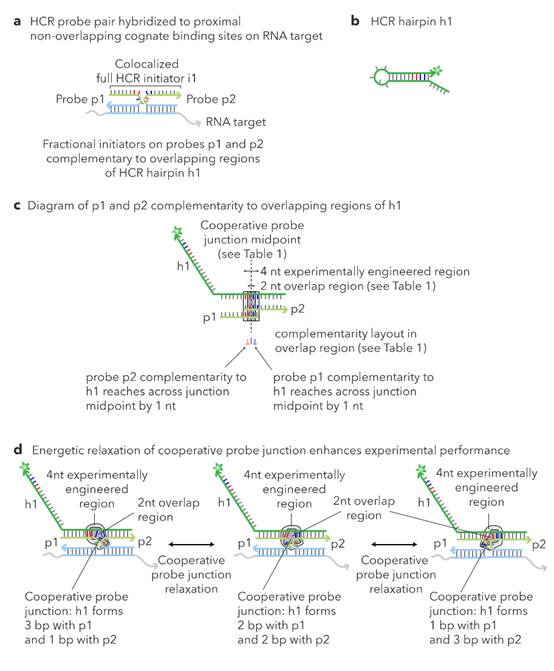
205. Essentially what happens is that the red nucleotides which are present in both p1 and p2 can compete to bind to the corresponding red nucleotides in h1; that is the overlap that MI relies on.
206. Because I found this difficult to make out from the PPD, I asked the parties to cooperate to agree a representation in which the overlap could be seen more clearly, with the identities of nucleotides marked, and to show, as well as the overlap case, what a situation would look like without an overlap.
207. This they did, and I am very grateful for what was produced. The precise length and the actual sequences of the overlaps in its commercial products are, understandably, maintained as confidential by MI, so the individual nucleobase identities shown in the diagrams are different from the real products (I asked MI to change them to bases which were complementary or not just as the case may be in their actual pairing, but which were different at the level of individual nucleobases, so for example it may have changed A-T to C-G).
208. The overlapping case looks like this:
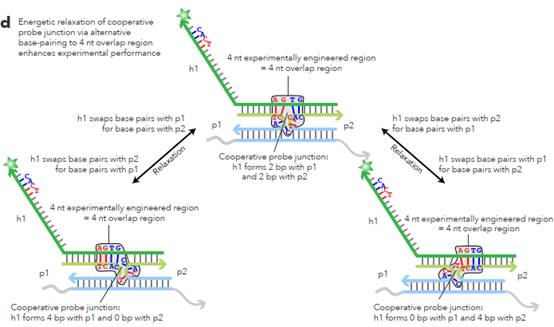
209. This shows the two ways in which the AG red nucleobases in h1 can hybridise to either of the instances of the corresponding TC, present in p1 and p2.
210. The non-overlapping case looks like this:
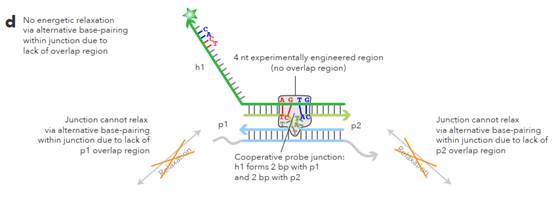
211. Here, there is only one way for h1 to hybridise to p1 and p2, and this means (MI says) there cannot be any "relaxation" of the junction, and the liability of the unzipping to get "stuck" remains.
212. MI's argument for non-infringement focuses on the overlap region where p1 and p2 meet and abut h1. ACD emphasises as part of its argument that despite this non-overlapping region, the outward parts of p1 and p2 (whose lateral extent was marked by ACD in pink, as shown below) will each hybridise to complementary regions in h1, and therefore still giving the advantage of using two probes rather than one, as discussed in EP572 at [0207]. The magnitude of this advantage, compared with having just one probe, may be reduced by the overlapping areas, but MI did not dispute that it exists. The PPD does not specify how long the shaded regions are, either in absolute terms or relative to the overlapping regions, but they can be seen schematically here (in pink, as I have said):
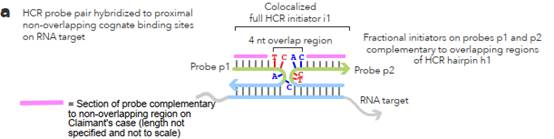
213. It is convenient at this point to deal with what the skilled person would expect, in the context of the way the Patents explain their invention, the effect of an overlap of the general kind used by MI, to be. I am, in this section, not dealing with the facts about MI's system in the real world (I resume that discussion when I come to the experiments, next), which would of course not be known to the reader of the Patents, but this topic is easier to follow here. Prof Tyagi drew out in schematic form what an arrangement according to the Patents would look like if there was an overlap (from paragraph 1.43 of his first report, based on Figure 4 of the Patents):
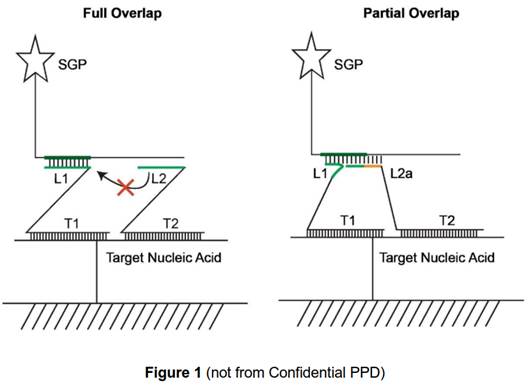
214. He explained that a partial overlap would be expected to lead to weaker binding (a complete overlap would prevent both probes binding and would be clearly undesirable). The partial overlap situation he showed is conceptually broadly equivalent to what MI do with their products.
215. Prof Tyagi expressed initial surprise that MI had managed to bring about a working overlap, but said that he concluded from the PPD that the energetic probe junction was relaxed as described above.
216. I did not however understand Prof Tyagi's written and oral evidence as going so far as to say that it would be expected that any overlap would entirely remove the advantage of having two probes rather than one. He just thought that having an overlap would reduce it.
217. Dr Wolf gave some vivid evidence that "nobody in its right mind designs probes that are overlapping unless you have very specific reason to doing that".
218. Understandably, MI relied heavily on this. But I do not think that Dr Wolf was saying that the skilled person's expectation would be that any degree of overlap would necessarily prevent a system from working, and she went on to give an example where overlapping probes were justified (an overlapping extension PCR technique). She was just saying that the natural inclination of the skilled person reading or implementing the Patents would be to have no overlap at all, and they would need a good reason to change from that. Once the skilled person was provoked to thinking about overlap my view is that they would expect that the benefit of two probes rather than one could be maintained with a degree of overlap, as long as there was good enough binding in the nonoverlapping part.
219. MI submitted extensive experimental evidence. ACD did not submit any experiments. It did not have any obligation to do so, of course, and in general it sought to make its case directly from the PPD. Its main attitude to the experiments was to dispute that they showed what MI argued for, although as a fallback it argued that so far as the experiments showed the advantage for which MI argued, it was only for a limited range of parameters and could be inferred to be absent for other sets of conditions, including ones of practical importance.
220. MI's experiments were not litigation experiments, but were conducted in the course of its business to make pragmatic decisions about how to configure its products, in particular in relation to the overlap in the probe pairs. As a result, they were not attended by the procedural regime of the Patents Court for litigation experiments. There was no notice of experiments, no formal list of facts to be proved and no opportunity for repeats. I do not say this by way of criticism (other than perhaps in respect of the facts that it was said were proved) but as a part of the context in which to assess their weight.
221. The experiments are also far short of what would be required for a peer-reviewed publication. Again, this is not a criticism as such, but part of the context, and MI did not submit that they were of that standard.
222. My function in this judgment is not to put myself in the position of a reviewer of a draft publication submitted to a scientific journal. I have no doubt that Dr Choi and Prof Pierce would never submit the experiments as they stand to a journal, and I also have no doubt that if they did, the journal would reject them. They have far too many issues to meet that exacting standard.
223. Instead, my function in this judgment is to make findings to the civil standard of proof about how MI's products work. I have to do the best with what I have before me.
224. It would have been helpful, in retrospect, to have had a formal statement of what MI said that the experiments proved. In essence, however, the question is whether they support the proposition set out in the last sentence of paragraph 2.22 of the PPD (which essentially describes the Alleged Improvement):
For fractional initiators with engineered overlaps, the cooperative probe junction is able to relax into an energetically more favorable subensemble of conformations to improve the strength of HCR signal amplification compared to not having the overlap.
225. In overview:
i) The experiments were of two kinds: in vitro gel studies, and in situ studies.
ii) The products ("HCR Amplifiers") tested were designated B1 to B20, but not all the products were tested in the in vitro studies or the in situ studies.
iii) Products B11 to B20 have never been commercialised, but the results on them still form part of the overall picture of what is going on, and ACD asked that I decide whether or not they would infringe, which I intend to do (on the basis that they exhibit the same behaviour as B6 to B10 in relation to the achievement of the Alleged Improvement).
iv) In the in vitro work, products B6 to B20 were tested, with a variety of overlap sequences, including in particular comparing various overlaps with non-overlapping arrangements.
v) In the in situ work, products B6 to B10 were tested (B11 to B20 never were) so as to compare non-overlapping and overlapping configurations.
vi) Also in the in situ work, products B1 to B5 were tested so as to compare different overlapping configurations, but with no comparison with non-overlapping configurations. MI does not rely on these.
226. The in vitro results have the following format:

227. The ON state and OFF state are explained as follows:

228. The "Target" values marked in red are for the non-overlapping configuration. All the subsequent "Target" rows designated in the same format (such as "Target +p1(u*==0)+p2(v*=1)" are overlapping configurations.
229. In the example given above, in the ON state it can be seen that the last row, marked in orange, has a better score than the non-overlapping configuration. It corresponds to the overlapping configuration specifically included for B6 in the PPD. It is not the only overlapping configuration that is better than the non-overlapping, and some overlapping configurations are less good than the non-overlapping.
230. This is obviously a complex picture, but the parties are agreed on the following point:
For each of the gel studies there is always an overlapping configuration [i.e. one or more] which has a higher conversion percentage than the non-overlapping configuration of probes for the same amplifier system.
231. The in situ results are given in the form of images, where the brightness of each pixel is processed. The following image was in the parties' agreed document on the experiments and although there are disagreements about interpretation I do not think there was any dispute preventing me from using it as an illustration at a general level:
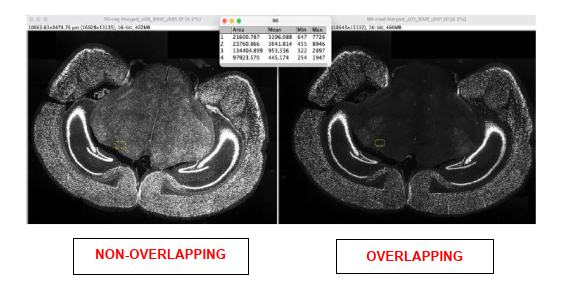
232. The following explanation of the interpretation of these results is taken from the parties' agreed document, but the parties were only agreed that it represents MI's position; ACD made clear that it did not accept it as a correct statement. However, I find that for my purposes at least it is a sufficiently accurate summary supported by sufficient acceptance on the part of Dr Wolf and evidence from Dr Choi (as a fact witness) and Prof Tyagi:

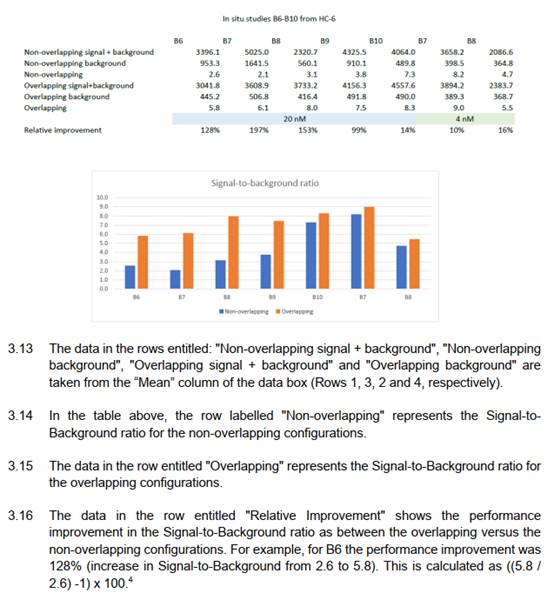
233. It should be noted that the various in situ experiments were done with differing probe concentrations.
234. MI summarised what was done for B6 to B10 in the following way in its closing written submissions:
339. The in situ studies for B6-B10 compared the nonoverlapping configuration to the overlapping configuration that had been judged to be the best in the gel studies. The results are contained in:
a. HC-3, at D5/3. These are each performed at 20nM.
b. HC-6, D5/6. This contains the same images as in D5/3, but additionally i) the "overlapping vs overlapping" results for B1-B5 at 20 nM and ii) repeats of the B7 and B8 studies, but at 4nM rather than the 20nM that was used for all other studies.
c. The quantification of the results is at XX-CW/12.
235. The parties also prepared a chart which helps to see what was done (this also covers the in vitro work):
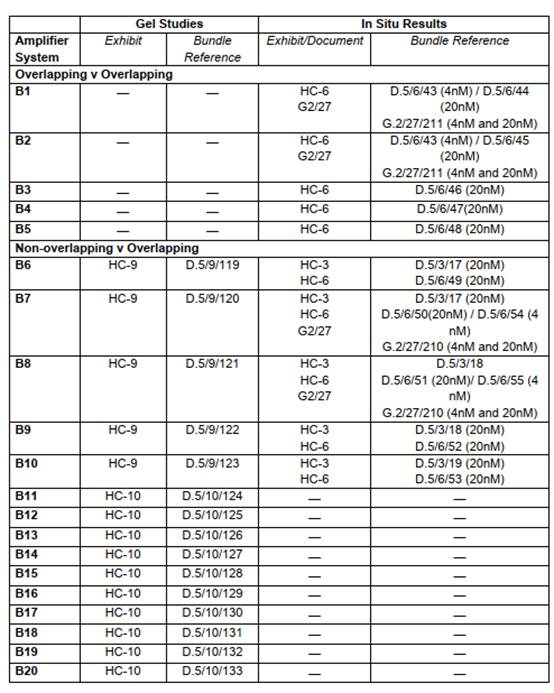
236. In closing, MI relied on the fact that Dr Wolf had said in her third report (at paragraph 3.20) that generally speaking the non-overlapping in situ images showed lower signal:background than the overlapping equivalents (which would imply that the Alleged Improvement was observed). However, that is not a complete statement of what she said in that paragraph, since she went on to say that the "brightness" on which this proposition might be based could be an artefact. She also went on to say that the data did not allow conclusions to be drawn, for reasons she elaborated. These were, she said, deficiencies in the experimental set-up and lack of appropriate controls.
237. MI also submitted that Dr Wolf changed her opinion during her oral evidence when she said that the overlapping and non-overlapping images looked equivalent. I think that submission was overstated; Dr Wolf was not changing her view from saying there was a difference to saying that there was not. She had said in her written evidence that there appeared visually to be a difference but that no conclusion could be drawn, and in her oral evidence was essentially saying that such difference as there was, was not material.
238. Prof Tyagi took the opposite view to Dr Wolf. He was not challenged on issues about appropriate controls and so I decline to attach weight to those points from Dr Wolf. He also was not challenged on a point made by Dr Wolf but which she backed down from in oral evidence about, she said, an inconsistency between the in vitro and in situ results.
239. One of Prof Tyagi's main points was that he found the quantification provided in XX-CW/12 important and convincing. Dr Wolf mainly questioned whether the very small number of data points at the 4nm probe concentration showed anything real.
240. As well as commending Dr Wolf's views over Prof Tyagi's in general, ACD made the following main points:
i) That MI's own personnel (Ms Ives and Dr Choi) differed in their conclusions about the experiments. I find this of little weight in itself and it is a second order consideration as I now have the assistance of independent experts. So far as it matters I prefer Dr Choi's views as somewhat more logical and in the light of his greater qualifications.
ii) That there was no improvement at 4nm probe concentration. I analyse this below.
iii) That so far as anything was shown, it was only at 20nm concentration which is not a concentration at which the MI products are explicitly instructed to be used. MI responded to this that users are recommended to use higher concentrations for lower expression targets and so they will use an appropriate concentration.
iv) That it is essential to have in situ results and that in their absence no conclusion can be drawn. Despite one encouraging answer (from ACD's point of view) from Prof Tyagi on this, I do not think he was addressing himself to the question of what can be concluded from the totality of the evidence. I return to this below.
v) That Prof Tyagi agreed that to understand properly any effect of the overlap would require full in situ data in a series of different models, and not just in mouse brains. I accept that but my task is not to decide if a full understanding exists; on the materials before me it plainly does not. Again, I return to this below.
241. I find it hard as a non-expert to judge matters directly from the images. So far as is relevant I share Dr Wolf's initial impression that the overlapping images look better. This favours MI, but only to a limited extent.
242. On the other hand, I find that the numerical analysis in XX-CW/12 is (more) objective and useful. It provides a significant showing of the Alleged Improvement.
243. On the 4nM point:
i) The MI internal views varied but for the little that it is worth, Dr Choi thought there was an effect.
ii) Prof Tyagi said in oral evidence that he thought there was no evidence of an effect at 4nM.
iii) MI meet that by saying that Prof Tyagi was not asked about or taken to the quantitative analysis in XX-CW/12 which does show an effect, albeit "much smaller" than at 20nM.
iv) MI also relies on the fact that Dr Wolf recognised that there is a small difference in the numbers, but accept that she said that as a single data point it was meaningless.
v) MI says that Dr Choi and Prof Pierce both pointed out that it would be expected that any effect would vary with concentration. As a matter of their status they were not giving evidence as experts, but rather as fact witnesses. However, the point makes sound sense and ACD has not really argued to the contrary.
244. Based on the in situ experiments as a whole I conclude that there is adequate albeit messy and not unproblematic evidence allowing me to conclude that the Alleged Improvement had a positive effect in those experiments comparing non-overlapping with overlapping at the 20nM concentration. The evidence at 4nM concentration is extremely weak; on its own I would not have been prepared to draw any conclusion at all from it, but in the context of the 20nM results and all the other evidence in the case I find on the balance of probabilities that while the Alleged Improvement is real, it is concentration dependent and in the test set-up used the effect dwindles to almost nothing at 4nM.
245. Plainly I could draw no conclusion in themselves from those instances (B1-B5) where no comparison of overlapping and non-overlapping configurations was done, but no reason was presented to think that they would have behaved differently. ACD just relied on the absence of in situ results for them.
246. As I have now explained, there was little dispute about the in vitro work and a good deal more disagreement about the in situ results. I have set out my assessment of the disputes on the in situ work above.
247. I consider the following matters to be important:
i) There is a theoretical basis why the Alleged Improvement may work: the energetic barrier discussed above. That this basis was plausible was not materially disputed by ACD.
ii) The theoretical basis cannot be pushed too far, though, because sometimes overlapping configurations are less good than overlapping ones (in the in vitro experiments).
iii) The theoretical basis is not, on the evidence before me, said to be able to predict which overlapping configuration should be used.
iv) The in vitro results show that the Alleged Improvement can be observed in all the HCR Amplifiers with at least one overlapping configuration, but the preferred configuration(s) differ from case to case.
v) The in situ results demonstrate the Alleged Improvement in some relatively limited situations.
248. In relation to the in situ results, I would also say that absence of evidence is not evidence of absence: even if the Alleged Improvement was not seen at some particular concentration (and I have concluded only by a narrow margin that it still occurs at 4nM but has dwindled almost to nothing then), that is not necessarily proof that it did not happen. The experiment is not set up to show that.
249. In my view I must take these matters in their totality. It would be wrong, for example, to decide the issue based on the theoretical basis alone (and I do not think I am asked to do so), not least because it clearly is an oversimplification and cannot predict or explain which overlapping configurations provide the Alleged Improvement and in what circumstances. On the other hand, it would be wrong to say that the in situ results on their own are weak, so discard them, and then move on to the in vitro results only to reject them for not being in situ (in any event I do not think the in situ results are as weak as that).
250. On the basis of looking at the three categories of evidence as a whole, I conclude that the Alleged Improvement is real and has a tangible effect in a range of practical circumstances. I say that because it is quite widely observed in the in vitro tests, is logical on the strength of the theoretical basis, and has adequate, if not especially strong, basis in the in situ results to think that it is not an effect which can only make itself felt in the in vitro environment for some reason. I also rely on the fact that ACD has not put forward a basis positively to think that if the effect were real in an in vitro setting, it would fail in situ.
251. A much more difficult issue would be identifying the full or precise range of circumstances in which the Alleged Improvement takes place. I do not believe I am in a position to do this, or indeed that I was asked to do so. For example, I am not able to say the lowest concentration at which it takes place, and other factors would also be at play, such as the rarity of the nucleic acid to be detected. Indeed, the concentration range over which MI's system is actually used was not explored in a lot of detail; the instructions refer to 16nM as an example of a "higher" concentration for single-molecule RNA imaging, but are not prescriptive. Although there was no evidence of 20nM (as used in the MI experiments) being used in practice I do not think it was unreasonable, and it was close enough to the 16nM mentioned in the instructions that I conclude it was representative of what would be likely to happen at that concentration in a broad sense. I do feel able to conclude that there is no positive reason to think that B11 to B20 would behave any differently from B6 to B10.
252. The fact that I am not able to identify the range of circumstances is of little practical importance however, since I conclude below that MI's system would infringe even given the existence of the Alleged Improvement, and that the Patents are invalid.
Claim scope issues
253. In this section I deal with "normal" purposive infringement and with equivalence. I deal first with an overarching point about the "non-overlapping regions" claim requirements, which is what the experiments were directed to, and then with a clutch of claim features which concern the labelling and amplification approach used by MI.
254. It is a requirement of claim 1 of EP572 (feature Eii)) and claim 1 of EP439 (feature F) that the L sections be complementary to non-overlapping regions of the label probe, amplifier, or pre-amplifier.
255. There is a matching requirement in each claim of the Patents that the T sections be complementary to non-overlapping regions of the target nucleic acid.
256. The claim feature refers to complementarity to the label probe, to the amplifier or to the pre-amplifier. I am going to conduct this part of my analysis by referring only to the label probe, for brevity. It does not affect the substance of my reasoning. I recognise that I also, separately, have to decide whether the h1 component is a "label probe" within the meaning of the claims (I address that below, concluding that it is). For the moment I call the h1 component a label probe for convenience only.
257. At this stage I am concerned with "normal" purposive interpretation and not with equivalence. It was not in dispute that this means addressing what would have been called purposive interpretation before Actavis v Lilly [2017] UKSC 48, except that equivalence does not enter into the analysis at this stage (see e.g. Icescape Ltd v Ice-World International BV [2018] EWCA Civ 2219 at [60]).
258. This is a case where it is necessary to see the disputed feature in the context of the whole claim.
259. Three basic points need to be observed so as to frame the right question.
260. First, the claim refers to "complementary", and this is a term defined by the specification (at [0081]) in terms which I have described above. It is a practical definition which depends on the functional ability of a polynucleotide as a whole to form a stable duplex with another polynucleotide. It is not a definition which works by looking at the level of individual nucleotides/nucleobases in the two polynucleotides and counting in how many pairs A goes with T or C with G; that is a legitimate way of describing relationships at the nucleotide/nucleobase level (see the ASCGK at 2.5) but not how the claims of the Patents work.
261. Second, the claim feature is not about whether the L sections overlap. This may seem obvious but MI's arguments tend to blur it.
262. Third, and a little more subtly, the claim requires that there are non-overlapping regions in the label probe to which each L section is "complementary", i.e. can hybridise to form a stable duplex. The claims call for looking for such regions. They are not addressed to identifying the whole parts of the label probe to which the L sections might hybridise and then asking whether those parts overlap.
263. I identify the relevant purpose as being that described in [0207] of EP572 - increasing specificity by having two probes so that non-specific binding is reduced, because if two probes hybridise the combination of L1, L2 and the label probe will be stable, but the hybridisation of a single probe will not. I do not think this was really in dispute.
264. Reference was also made to [0102] of EP572. ACD's argument was that if MI's construction of this claim feature were correct then "distinct" and "non-overlapping" would mean the same thing. MI accepted that that was the consequence of its position but said that "distinct, non-overlapping" was just the use of two words to give more emphasis to the same concept. There is something to be said for both readings, but I think each involves over-meticulous verbal analysis and even if that were not so, the weight that I would give the point is small. This is not, after all, an argument about whether a claim includes redundant wording, but over the precise relationship of very similar words in a general, descriptive part of the specification. For what it is worth, I prefer ACD's position.
265. All that being so, I can turn to consider the meaning of the claim feature and how it relates to the alleged infringement.
266. In my view, the claim feature just requires one to look at the L sections and ask if, for each, there is a region on the label probe with which it is capable of forming a stable duplex, with the two regions not overlapping. Put in terms of the marked-up figure submitted by the parties, the pink regions identified by ACD do this, and MI does not dispute that this means that its products achieve the kind of benefit stated in [0207].
267. This approach to claim construction means that the complexities of what happens in and with the "overlap region" in the MI product are just not relevant. I think this makes sense.
268. First, the overlap between p1 and p2 (4 nucleotides in the figure) is not what the claim feature is about at all; they are an overlap of the L sections and the claim is about non-overlapping regions of the label probe.
269. Second, while (1) p2 is capable of hybridising to the right hand pink portion plus the four nucleotide section on the label probe opposite the cooperative probe junction, and (2) p1 is capable of hybridising to the left hand pink section plus the same four nucleotide section, and (3) those two sections of the label probe overlap in the middle, that is a different question and it would seem to the skilled person that the overlap did not undercut the purpose of the invention, if, as is the case, there was adequate hybridisation to the pink regions.
270. There is no doubt that the skilled person reading the Patents would most naturally think of a set-up where the L sections in their entirety would predictably hybridise to parts of the label probe which did not overlap, as is exemplified in figure 4 (they would, reading [0102] also think that the best approach was for those two regions of the label probe to be contiguous). I think that is really just another way of saying that they would not in practice spontaneously turn their minds to the issue of claim interpretation that I have to consider.
271. What of the experts' evidence on the wisdom of having an overlap? I have addressed this above. By "overlap" now, I mean in the sense of the 4 nucleotides in the label probe to which p1 and p2 compete to bind. As I have said above, the upshot of that evidence is that the competition would not, in itself and all other things being equal, be a good thing. It could be a bad thing to the extent that it reduced the confidence that each L section would bind stably, and it would definitely be a bad thing if it was so large that it prevented the formation of two stable duplexes, but then it would be outside the claim on my analysis anyway. But if the skilled person were aware that there were parts of each of the L sections that could stably bind to the label probe, then I do not see that they would have the same problem with the "overlap", especially if there were some case-specific positive reason for having it.
272. In case I am wrong in my analysis so far, I will consider matters on the basis that the correct approach is to identify the region defined by the greatest extent of the label probe to which each L section is capable of binding, and then to see if those overlap. In terms of the parties' figure, those regions are the ones I have identified above (for p1 the left hand pink section plus the four nucleotide section in the middle, and for p2 the right hand pink section plus the same four nucleotides). These therefore overlap in the four nucleotide stretch.
273. From this perspective, the relevant question would be: does "non-overlapping" mean "completely non-overlapping"? If this were the right question, I think the skilled person's reaction would be much as above from a practical point of view: the simplest approach would be to have no overlap (of this kind) at all, but that so long as there was enough non-overlapping sequence on each side of the label probe beyond the overlap to allow two stable duplexes to form, the invention would work and the key benefit would be achieved.
274. I find a (slightly loose) analogy to Catnic helpful. There, the relevant integer required a weight-bearing part of the claimed product to be "vertical". A small degree of variation from vertical was within the claim, purposively interpreted, because the skilled person would know that the basic weight bearing capability was preserved, only reduced to a modest degree not impeding its function. So here: "non-overlapping" need not mean "completely non-overlapping". The analogy breaks down somewhat because in Catnic it was possible to tell that the angle of deviation from the vertical was immaterial by simple trigonometry, whereas in the present case it is more empirical. But the question is still quite a simple one: whether the two probes each form stable duplexes so as to get the benefit of two probes rather than one. There was no allegation of insufficiency that it was not possible to tell whether this would be so.
275. ACD relied on the process by which MI got to its setup: it aimed to have no overlap (i.e. for the type of separation indicated in figure 4 of the Patents) but found, serendipitously, that a small overlap solved the problem of the energetic barrier presented by the cooperative probe junction, that problem itself stemming from the particular hairpin arrangement of the h1 component and the interaction of the probes used.
276. I do not think it can be directly relevant to infringement how the defendant arrived at its method, which either infringes or does not. But it does illustrate the overall sense of the approach of the skilled person for which ACD contends and which I have found in favour of: the key thing is to form two stable duplexes, and while the simplest way to achieve that is to have no overlap, it may be all right and indeed necessary to have a small overlap if there is a positive reason for it, and the key goal is not lost.
277. This conclusion means that it is not necessary to consider infringement by equivalence separately, but I shall do so briefly. I need to do so on the assumption that I am wrong about purposive, "normal" interpretation, which I confess I find involves a more than usual degree of mental gymnastics.
278. I consider that the "inventive core" (as explained in Icescape, supra) is that set out above by reference to [0207] - better specificity by the use of two probes where one would bind unstably (in an in situ assay). For reasons I will come to below, I do not think the inventive concept includes the amplification system, but that is a separate matter.
279. That helps identify the relevant level of generality, which is a key issue in answering the Actavis v Lilly questions, to which I now turn.
280. The first question is whether the variant achieves substantially the same result in substantially the same way. In my view the answer is "yes", since it is not disputed that two stable duplexes are formed by two probes and that the benefit of doing so in terms of specificity is achieved. The result is the good specificity and the way is two stable duplexes.
281. MI submitted that based on the experiments the answer was "no" because what it called the "overlapping" configuration was better than the "non-overlapping". But I think this approaches matters at the wrong level of generality.
282. The second question is whether it would be obvious to the skilled person, knowing that the variant achieved substantially the same result, that it does so in substantially the same way? Given the answer to the first question, this can only be answered "yes", since if the skilled person knew how the variant worked they would also immediately see how.
283. The third question is whether the skilled person would have concluded that the patentee nonetheless intended that strict compliance with the literal meaning of the relevant claim(s) of the patent was an essential requirement of the invention. I do not consider there is anything in the specification to support the patentee having that intention. MI made two points, however:
i) First, it said that the non-overlapping feature was "an essential part of the inventive concept o[n] both parties' cases". That adds nothing to what I have said above in accepting ACD's case that what matters is two stable duplexes.
ii) Second, it said that the feature had technical significance, since the CGK comes into play in question 3 (which I accept for present purposes), and would lead the skilled person to conclude that having overlapping sequences was foolish. This fails on the facts in any event, since I have found that the skilled person would think that the most straightforward approach was to have no overlap but that some overlap would be all right.
284. Three points arise on this issue.
285. First, does the label probe have to hybridize directly to the capture probes?
286. If it does, then ACD's case would have to be that the first h1 unit added, and only that, was the "label probe" of the claims, and this leads to the second and third issues:
i) The second issue is whether a "label probe" of the claims has to be such that it, on its own, "enables" the target to be detected. MI says that it does but that the label on the first h1 is not good enough for that purpose.
ii) The third issue is whether the label probe has to be a terminating molecule.
287. But on the other hand, if the label probe does not have to hybridize directly to the capture probes then the claim feature could be met by a concatenation of h1 and h2 units, including the terminating molecule.
288. I will deal with the second and third issues first.
289. I observe as a general matter that all the claim language relating to the label probe, amplifier and pre-amplifier, and the definitions in [0083], [0085], and [0086], is broad and permissive. The very fact that there are three options in the claim (label probe, label probe with amplifier, label probe with pre-amplifier and amplifier) speaks to the patentee's intention to claim broadly. This is also consistent with the view that I have expressed above that the real inventive concept of the claims lies in the use of two probes. Labelling and detection are necessary to exploit the inventive concept, but the details would not be regarded by the reader of the Patents as being critical.
290. I also observe that while the skilled person would know of bDNA from their CGK to the extent indicated above and would in any event read of it in the Patents' specification, the claims are plainly not limited to bDNA (since e.g. no amplifier is required by them) and the skilled person would not think that they were. I reject MI's (rather faint) suggestion that they would. They would, on the other hand, think that bDNA was one way of implementing the Patents.
291. Finally in relation to general matters, I consider that "label probe" being a defined term makes it relatively more unlikely that there are requirements of the claim feature other than those stated in the definition.
292. MI bases this argument on the words "enables the target to be detected" in the definition of "label probe" at [0083]. There are two parts to it: the construction question and (when it comes to infringement) a factual question of whether the label on the first h1 in the MI set-up would be detectable on its own.
293. This argument was at best faintly made in MI's opening submissions and certainly not in the way that it is now.
294. As well as the general broad intent that I have referred to above, the use of the words "directly or indirectly" in [0083] militates against MI's restrictive approach, and in any event the word "enables" does not in ordinary usage necessarily mean something enables all on its own; it can perfectly well extend to something that makes a contribution towards a result.
295. Also, and I think decisively, the figures of the Patents - figure 8a is an example - have arrangements where there are multiple, numerous label probes but the skilled person's reaction would be that no one of them on its own would necessarily be detectable. So I do not think it is a requirement of the claims that the label probe has to be detectable on its own.
296. As to the facts, I do not in any event think that Dr Wolf accepted that the first h1 label on its own in the MI arrangement could not be detectable. All she accepted was that a single h1 label in a concatenation would be likely to give the same signal as if there was only one h1 present, and that the overall signal in the MI set-up is an additive combination of all the hairpins polymerised together. It may, in principle, be the case that the first h1 label on its own would not be detectable if the same experimental set-up as for polymerised h1s and h2s was used but in the circumstances it was for MI to raise the point much more clearly and to prove it, and the evidence which it has is not good enough, I find.
297. MI submits that the label probe has to be a terminating molecule, and the support for this contention is said to be:
i) That there is nothing in the CGK where a label probe is onwardly bound to something else, and in bDNA in particular labels are not onwardly bound but are the terminating molecule. As to the CGK point there is no reason to suppose that the patentee wanted to limit themselves to the CGK and every reason to think they did not, in the light of the point above about the inventive concept and the broad intent in relation to the labelling and amplification. The fact that bDNA has the labels as terminating molecules is a very similar point conceptually and rather amounts to saying that the skilled reader would think the claims were limited to a specific CGK arrangement. It is illegitimate to read general words as limited to something specific in this way.
ii) That where the Patents describe a molecule which is an intermediate, they say so, as in [0198] referring to the amplifier. However, the amplifier needs to be an intermediate to serve its function so it is not surprising that the Patents stipulate this. It is a non-sequitur to say that because the patent is explicit when something has to be an intermediate, when it is silent then the thing in question cannot be an intermediate.
iii) That [0198] at lines 21-25 says ""... In indirect mode, the label probe can either be attached to the target molecule through binding to a capture probe directly or through binding to an amplifier that is in turn linked to a capture probe...". MI submits that this is a limiting list and that there is no possibility of the label probe being bound to another label probe. This is not logical, either. The quoted text is about how the label probe is attached on the target side, and says that the attachment can be direct or indirect. On this part of ACD's argument, the label probe is directly attached and that is specifically allowed by the language. The language is not about any attachment on the other side of the label probe.
iv) That all the figures of the Patents have the label probe as the terminating molecule. This is true (and I reject any suggestion by ACD that Figure 8b is an exception, since although it has a concatenation of SGPs the label probe is the terminating molecule) but it is not legitimate to read general claim wording down to the preferred embodiments in this way.
298. So I do not think there is anything in any of MI's points and had the patentee intended to require a label probe to be a terminating molecule it would have been done by saying so in the definition. If anything, the definition of "label probe" at [0083] refutes MI's argument when it says at line 25 that it "directly or indirectly" provides a detectable signal.
299. Since I agree with ACD on the second and third points, it does not matter whether the label probe has to hybridise directly to the capture probe; even if it does, the first h1 fulfils the requirements to be a "label probe", which is in any event what ACD argued. A closely related point about direct hybridisation arises in relation to the "amplifier" issues, however, and I deal with it there.
300. The central debate and the quandary in which MI says that ACD is caught can be captured by the following figures from Prof Tyagi's evidence with the explanatory text which goes with them:
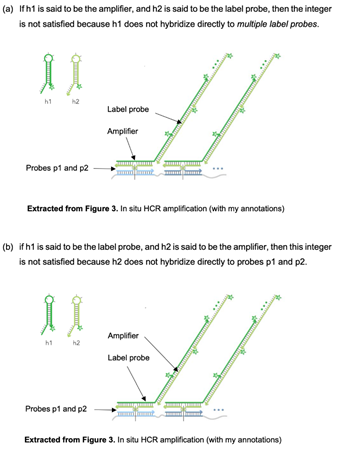
301. Thus MI submits that whether ACD takes h1 or h2 as the "amplifier" there is no infringement. In each case, in different ways, the point depends on the claim requiring that hybridisation has to be direct, which MI says is the correct approach given the definitions in [0078] (of "hybridize") and [0085] (of "amplifier").
302. ACD's case, at least by closing oral submissions, was that the amplifier called for by the claims is the first h1 (the one hybridized to the capture probes) and the multiple label probes are h2. It accepted, I think, that that particular h1 only hybridizes directly to one h2, although it emphasises that any given h1 is capable of hybridising directly to two h2s, and later h1s in the chain will do so.
303. Since I have found that ACD succeeds on its first argument, based on option (i) in the claims (first h1 as a label probe), I will be brief on this point.
304. First, I consider that ACD succeeds on the basis that each h1 is capable of hybridising to multiple h2s and that is what the claims call for in respect of the amplifier.
305. Second, although the definition of "hybridize" in [0078] in a literal sense might be taken to exclude indirect attachment, what it is really stressing is that the attachment is by nucleic acid hybridisation (rather than e.g. covalent attachment) and that the association is strong enough to form a stable duplex. MI castigated ACD for relying on "indirect hybridisation", and said there was no such thing. That is in a sense true as a matter of molecular biology, but ACD was not relying on a different kind of attachment at the molecular level, it was focussing on a multi-stage attachment in which each stage in the chain of overlapping h1-h2-h1-h2s etc was by hybridisation in the normal way.
306. Third, the language used in describing the "amplifier" at [0085] is broad and inclusive, indicated by the repeated "typically"s.
307. Fourth, the skilled person would understand that what was important was the attachment of an amplifier and then, downstream from it, multiple labels. No importance is attached at a linguistic level to the morphology/topology of the amplifier/labels complex.
308. I think these are all significant points in ACD's favour and I think MI's approach is excessively literal and linguistic. I could not see any practical points in MI's favour.
309. Had I concluded that MI was right on purposive interpretation, I would have had to consider equivalence. My assumption to do that would have had to be that the purposive interpretation required one amplifier to be attached directly to multiple labels and that the variant was that the multiple labels were attached indirectly, by succeeding hybridisations, in a concatenated rather than branching way.
310. My analysis of the equivalence allegation would have been informed by my view of the inventive core of the Patents, set out above. That is all to do with the use of two capture probes, as I articulated at greater length there. The claims do ultimately require a strong enough signal to be capable of detection in some way, but options (i), (ii) and (iii) all provide it, in different ways.
311. On question 1 I would have decided that the variant made no difference to the way the invention worked. The use of two capture probes is unchanged and a strong enough signal continues to be provided. Even if I thought the inventive core was in some way more specific I would not have held that it resided in, specifically, a branched amplifier arrangement. The result is the same, and certainly substantially the same.
312. Question 2 adds nothing, once question 1 is answered in ACD's favour.
313. On question 3 I would have held that there is nothing in the specification to indicate that direct hybridisation was a strict requirement. MI's written submissions were directed to the proposition that the specification requires multiple label probes for each amplifier. I do not think that captures the right question. ACD's argument does not exclude there being multiple label probes for each amplifier, but it does involve "indirect" attachment. There is nothing in the specification to exclude that.
314. As I have set out when explaining the issues at the start of this judgment, I proceed for the purposes of this trial on the basis that title to MI's products passes to its customers in the USA and that MI does not itself commit a relevant act in the UK. My decision at this trial is limited to whether MI is jointly liable with its customers in respect of their acts of importation or use.
315. Importation would be the relevant act in relation to EP439 with its product claims; use is the relevant act in relation to EP572, which has method claims.
316. However, the claims of EP439 are to kits with a permeabilising agent and no permeabilising agent is included when the MI products are imported. So even if MI were jointly liable for the importation, it would not amount to an infringement. ACD had no answer to that, and Counsel for ACD effectively abandoned this part of its case in his oral closing. The contact with its customers that accompanies the importation could in principle, I think, still have formed part of the overall picture going to joint liability, even in relation to customers' use of the MI products, but in practice the evidence was in two separate streams (Ms Pierce speaking to importation and Dr Choi almost entirely to use, although he touched very slightly on importation) so I do not think I need say any more about importation.
317. The parties were agreed that the relevant principles are to be found in Fish & Fish Ltd. v. Sea Shepherd UK [2015] UKSC 10. The parties also agreed that although the Justices of the Supreme Court in that case divided 3:2 on the result of the appeal, that was because of a different view of the facts and not the law. I accept what the parties say both in relation to its being the relevant authority and as to the fact that the Supreme Court was in agreement on the law.
318. I do not think I need to find help in authorities before Sea Shepherd and one has to be careful about them, because notions in them such as the defendant "making the infringing acts its own" are deprecated in it, and because the Supreme Court counselled against any gloss on the standards it set out (see below).
319. I also do not find it helpful or appropriate to compare facts between the present action and the authorities, especially authorities before Sea Shepherd. I think I should identify the relevant principles and apply them.
320. I was referred to what Lord Toulson said at [21]:
To establish accessory liability in tort it is not enough to show that D did acts which facilitated P's commission of the tort. D will be jointly liable with P if they combined to do or secure the doing of acts which constituted a tort. This requires proof of two elements. D must have acted in a way which furthered the commission of the tort by P; and D must have done so in pursuance of a common design to do or secure the doing of the acts which constituted the tort. I do not consider it necessary or desirable to gloss the principle further.
321. To what Lord Sumption said at [37]:
...the defendant will be liable as a joint tortfeasor if (i) he has assisted the commission of the tort by another person, (ii) pursuant to a common design with that person, (iii) to do an act which is, or turns out to be, tortious.
322. And to what Lord Neuberger said at [55]:
It seems to me that, in order for the defendant to be liable to the claimant in such circumstances, three conditions must be satisfied. First, the defendant must have assisted the commission of an act by the primary tortfeasor; secondly, the assistance must have been pursuant to a common design on the part of the defendant and the primary tortfeasor that the act be committed; and, thirdly, the act must constitute a tort as against the claimant. As Lord Toulson says, this analysis is accurately reflected in the statement of the law in Clerk and Lindsell on Torts, 7th ed, p 59, cited by all members of the Court of Appeal in The Koursk [1924] P 140, 151, 156, 159.
323. Sea Shepherd has been considered in numerous cases since. I was referred to the decision of the Court of Appeal in Glaxo Wellcome v Sandoz [2017] ECWA Civ 227 (a passing off case), but to my mind that just confirms that the Supreme Court was divided as to the facts and not the law, and refers to all three of the formulations I have identified above.
324. I think that the formulations above are to the same effect, just phrased differently. Personally, I find Lord Toulson's expression particularly useful because it makes explicit the point that "mere" facilitation is not enough.
325. MI provides customers with general and standard directions as to the use of its products by way of protocols. Were this all that happened then I would conclude that MI had gone no further than mere facilitation.
326. However, in relation to some customers, MI becomes more directly involved. This happens when customers need help, in which case MI provides "troubleshooting". A number of examples were taken up with Dr Choi in his oral evidence, his having been personally involved in such activities at an earlier time in MI's history when the company was much smaller and he had direct customer contact at the same time as being CEO.
327. When this sort of situation arose, Dr Choi would provide tailored advice and tips direct to customers for their specific needs and problems. An example when he proposed to a customer "let's troubleshoot together" captured the flavour of this sort of interaction. MI, through Dr Choi, would not only know the exact parameters being used by customers to get the products working, but would steer and recommend the choice of those parameters. This assistance was rendered in the context of a common design to get the products working by a method which, on the conclusions I have reached on claim scope, would have infringed had the Patents been valid.
328. MI submitted that:
i) The interactions between it and its customers were "unremarkable". I agree but it is nothing to the point. No doubt this fact pattern is common and many suppliers of bespoke equipment which needs detailed dialogue and advice for proper use would be jointly liable with their customers. That is simply because those situations would be likely to meet the legal standard.
ii) The customers initiated the contact when troubleshooting was needed. Again, this is true but it cannot help MI if what happened thereafter amounted to a common design in law.
iii) That products often come with instructions. I agree and as I have said above, if that were all then I would not have found in ACD's favour. MI sought to draw an analogy to Sabaf v Meneghetti [2002] EWCA Civ 976. I have rejected this sort of fact comparison above, but in any event that was just a case where it was decided that merely acting as a supplier was not enough.
iv) That MI did not have a contractual obligation to provide support. Again, this is true and it could perhaps provide a part of the factual picture in some cases. But it cannot be a requirement of joint liability that the person alleged to have been the joint tortfeasor was contractually obliged to do what it did. In my view the culpability and the legal basis for joint liability could perfectly well apply if the joint tortfeasor acted voluntarily but out of self-interest, as MI did. I make clear that there was nothing immoral in MI's helping its customers; on the contrary it was an entirely decent and understandable thing to do. MI provided no authority in support of this argument.
v) That there are many MI customers who do not require support of this kind. I agree with this. Had the Patents been valid, and depending on the result of the deferred question of where title passes, this might have led to a difficult decision in relation to relief, but it would not mean that MI was not liable where it did provide such support.
329. I therefore reject MI's points and conclude that it would have been jointly liable in respect of the above kinds of act of bespoke troubleshooting had the Patents (specifically, for reasons given above, EP572) been valid.
330. In fairness to MI, I should record that the argument on behalf of ACD which has succeeded was only added, or at least identified with any clarity, by an amendment to its pleadings at trial.
331. MI's main focus of attack was over the prior art, based on Collins, either alone or with Kern. Its secondary prior art case was over Player, and insufficiency and added matter were only squeezes.
332. There was no material dispute about the overarching standard: there has to be a disclosure of clear and unmistakable directions which, if performed, would result in something necessarily falling within the claims. It is possible for such a disclosure to be implicit rather than explicit, and I do not think that was in dispute, but one has to be very careful then not to veer into the territory of obviousness.
333. It is also necessary that the disclosure be enabling.
334. There were two possible areas of dispute about the law. One was in relation to combining documents ("mosaicking") and the other was in relation to prior art which discloses a number of options. These arose because, first, MI says that Collins may be combined with Kern for the purposes of novelty and, second, Collins discloses a number of options in the key passage. As it happens, I do not think ACD really made a point about there being multiple options, but I will touch on it anyway.
335. As to mosaicking, it is wrong to say that there is an absolute rule against combining documents for the purposes of anticipation. See Terrell on the Law of Patents, 19th Ed., 11-61 to 11-63. But it only becomes possible if one document points to another, and MI did not argue other than that the pointer has to be to the overall standard of clear and unmistakable directions, which I believe must be correct. Similarly, the fact that there is a general cross-reference from document A to document B does not of itself entitle a party attacking a patent to pick and choose anything from the disclosure of document B to add to document A, any more than it would be permitted to combine parts of document A if there were no disclosure, to the necessary standard, to do so (see Terrell at 11-63). One has to ask to what, in document B, there is a clear and unmistakable pointer. In the end, I do not think MI went any further than this.
336. As to disclosure of options, while a generic disclosure of a class does not destroy the novelty of a particular member of the class, individually identified members of a group of options can. See Terrell at 11-64 to 11-66.
337. In its written opening submissions, ACD made a somewhat complex argument about the relationship between the rules on combining documents as they relate to anticipation, obviousness and insufficiency. I do not find this comparative exercise helpful or necessary; I have set out the anticipation standard above, ACD accepts that it was obvious to consider Collins and Kern together for the purpose of obviousness, and there can be no dispute that when it comes to sufficiency the question is a completely different one because the skilled person has the disclosure of the patent in question, including the claims, so that they know what to aim for. I also found ACD's analysis a little unsatisfactory because it tended to suggest that when dealing with obviousness there is a different approach to the disclosure of the prior art than when dealing with anticipation. The correct approach, I consider, is to decide first what the prior art discloses, and then to ask what, if anything the disclosure renders obvious. But this is of little practical importance in the present case given ACD's acceptance that in relation to the obviousness attack, given the disclosure of Collins as it concedes it to be, it would be uninventive for the skilled person to consult Kern and to have the relevant idea of the claims of the Patents, the issue being only the assessment of the likely prospects of success.
338. The basic approach is as set out in the decision of the Supreme Court in Actavis v. ICOS [2019] UKSC at [52] - [73], with its endorsement at [62] of the statement of Kitchin J, as he then was, in Generics v. Lundbeck [2007] EWHC 1040 (Pat) at [72]. There was no dispute about this.
339. Collins was published in 1997 and its authors were mainly from Chiron with the exception of the last-named author who was from the Aaron Diamond AIDS Research Center in New York.
340. The title is "A branched DNA signal amplification assay for quantification of nucleic acid targets below 100 molecules/ml".
341. The Abstract is as follows:
ABSTRACT
The branched DNA hybridization assay has been improved by the inclusion of the novel nucleotides, isoC and isoG, in the amplification sequences to prevent non-specific hybridization. The novel isoC, isoG-containing amplification sequences have no detectable interaction with any natural DNA sequence. The control of non-specific hybridization in turn permits increased signal amplification. Addition of a 14 site preamplifier was found to increase the signal/noise ratio 8-fold. A set of 74 oligonucleotide probes was designed to the consensus HIV POL sequence. The detection limit of this new HIV branched DNA amplifier assay was -50 molecules/ml. The assay was used to measure viral load in 87 plasma samples of HIV- infected patients on triple drug therapy whose RNA titers were <500 molecules/ml. In all 11 patients viral load eventually declined to below the detection limit with the new assay.
342. As is touched on in the ASCGK/technical section above, isoC and isoG will pair to each other but not to the respective natural nucleotides, so non-specific binding is reduced.
343. The Introduction is as follows:
INTRODUCTION
Quantitative hybridization assays based on branched DNA signal amplification are widely used to monitor patients on antiviral therapy for human immunodeficiency virus (HIV), hepatitis C (HCV) and hepatitis B (HBV) and to stratify patients for therapy (1–7). They have also been used to predict time to onset of AIDS (8–10) and to establish the kinetics of HIV production and destruction, which has led to new insights into the mechanisms of pathogenesis (11,12). The most important characteristics of these hybridization assays are sensitivity, wide dynamic range, and precise and accurate quantification. The branched DNA hybridization assay pictured in Figure 1 employs linear signal amplification rather than exponential amplification of target.
A family of oligonucleotides called capture extenders (CEs) is used to capture the target to the solid support. The target is labeled by virtue of binding a large number (typically >30) of target-specific oligonucleotides called label extenders (LEs). In the first generation assay the LE probes bind a branched DNA amplifier (bDNA), which in turn binds many alkaline phosphatase probes. In the second and third generation assays, the LE probes bind pre-amplifiers, which in turn bind many amplifiers. The result is stronger signal amplification and lower detection limits. In all versions of the assay, the linearly amplified signal is directly related to the number of targets present in the original sample. This first generation bDNA assay has been shown to quantify nucleic acid targets accurately and precisely between -10 000 and 10 000 000 molecules; assays for HIV, HCV and HBV have been developed (13–17). The second generation HIV bDNA assay has a quantitative detection limit of 500 molecules (18).
The recent introduction of HIV protease inhibitors has driven viral loads to below even the detection limits of the second generation HIV bDNA assay (1,2). The first and second generations of the assay were limited by non-specific hybridization (NSH) between the amplification sequences and other nucleic acids. Short regions of hybridization between any member of the amplification system (alkaline phosphatase probe, amplifier or preamplifier) and any non-target nucleic acid sequence will lead to amplification of background. Capture probes (CPs), CEs and sample nucleic acids are all sources of this background hybridization. The purpose of this study was to examine the effect of redesigning the amplification molecules by incorporation of the non-natural bases, isocytidine and isoguanosine, to reduce their hybridization potential to all non-target nucleic acids. If target-specific signal amplification is accomplished without a concomitant amplification of the background from non-target molecules, then sensitivity should be greatly improved.
344. Figure 1, which, as explained, shows details of the various "generations", is as follows:
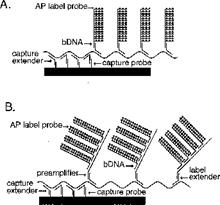
Figure 1. Basic bDNA assay components. (A) First generation assay; (B) second and third generation assays. The preamplifier (heavy lines) is unique to the second and third generation assays.
345. Kern is reference (18) in Collins and is referred to on a number of occasions, including in the Materials and Methods section.
346. Detection limits are given in the Results section on page 2982 and then there is the following:
Utility of system 8 bDNA assay in monitoring HIV-infected patients on triple drug therapy
The system 8 bDNA assay was used to measure viral load in 87 samples from HIV-infected patients on various therapy regimens, including triple drug therapy consisting of Nelfinavir, AZT and 3TC. Figure 2 summarizes the results of this study.
The viral load of the 11 patients showed a nearly monotonic decline to below the assay detection limit during the treatment. A total of 47 samples (54%) quantified above the detection limit and 78 samples (90%) quantified above zero. Only the development of a still more sensitive assay will be able to validate the positivity of the 31 samples quantifying between 0 and the detection limit. In spite of the fact that the average sample contained -130 virions/ml, the average CV (of the RLU) of the duplicate samples was only 14%.
347. This implies the need for greater sensitivity.
348. The passage which attracted the greatest focus at trial and which is the central basis for MI's attack, is the first four paragraphs of the discussion:
DISCUSSION
A more sensitive branched DNA assay has been developed to quantify nucleic acid targets <100 molecules/ml. This system 8 bDNA assay employs isoC and isoG nucleotides in the amplifica¬tion molecules to reduce backgrounds and allow for stronger amplification. System 8 amplification was used to show that triple therapy is at least eight times more effective (<60 molecules/ml) than determined using the previous version of the branched DNA assay (<500 molecules/ml). If 10 ml samples were concentrated before the assay, the assay could theoretically detect ~6 HIV RNA/ml or 3 virions/ml.
In theory the system 8 bDNA assay can be made considerably more sensitive not only by increasing volume, but also by increasing the S/N ratio. Most of the background is coming from LE NSB and amplifier NSB (data not shown). By using cruciform LEs, a design in which two LE probes must bind the target in the correct orientation to bridge the preamplifier, most of the LE NSB can in theory be removed (18). By finding more effective blockers for the solid phase or by redesigning the branched DNA molecule or the solid phase itself, much of the amplifier's NSB can be removed. Alternatively, by using sequences of reduced complexity (such as trinucleotide repeats), lower concentrations of the amplification molecules can be used during hybridization, resulting in reduced NSB. Additional layers of amplification can also be added.
A prototype system 8 bDNA assay has also been developed to quantify HCV RNA in plasma. A total of 12 2'-O-methyl probes (eight LE and four CE) were designed to the well-conserved 5' untranslated region of the genome. The assay has a detection limit of 200 molecules (unpublished data), which is ~50-fold better than the HCV 2.0 QuantiplexTM assay (15).
The system 8 bDNA assay should also be useful in other hybridization assays. In both filter and in situ hybridization assays, for example, billions of overlapping, unique oligonucleotide sequences are available for possible NSH to probes. The ability to amplify the signal without amplifying noise from hybridization of amplification molecules to sample nucleic acid sequences should greatly improve the sensitivity of these assays. Currently, in situ PCR is the standard for detection of single copy DNA sequences in cells (28,29); in situ RT–PCR has occasionally been problematic for mRNA detection (30–33). RNA targets that are partially degraded or intramolecularly crosslinked at selected sites should pose no special problems for in situ bDNA assays since priming and reverse transcription are not required. As in assays that target DNA, multiple oligonucleotides will be used to label target RNA in cells; failure to bind one or more of these oligonucleotides is of no real consequence. Quantification may also be possible with the in situ bDNA assay with proper selection of internal standards. The sensitivity of the system 8 bDNA filter and in situ hybridization assays should be limited mostly by the specificity of the oligonucleotide probes. Empirical selection of the best LE oligonucleotide probes and the use of the cruciform design (18) should prove most useful in optimizing specificity.
349. Kern is an article in the 1996 edition of the Journal of Clinical Microbiology. Its title is "An Enhanced-Sensitivity Branched-DNA Assay for Quantification of
Human Immunodeficiency Virus Type 1 RNA in Plasma". Its authors are again mainly from Chiron, with one from Agouron Pharmaceuticals. The abstract is as follows:
The quantification of human immunodeficiency virus type 1 (HIV-1) RNA has facilitated clinical research and expedited the development of antiretroviral drugs. The branched-DNA (bDNA) assay provides a reliable method for the quantification of HIV-1 RNA in human plasma and is considered one of the most reproducible assays ready for use in clinical trials. A series of oligonucleotide probe design and solution changes have been developed to enhance the sensitivity of the bDNA assay while maintaining its performance characteristics. Among the changes incorporated into the enhanced-sensitivity bDNA (ES bDNA) assay to reduce the background level and enhance the signal are the use of shorter overhang sequences of target probes for capture, the cruciform design of target probes for amplification, and the addition of preamplifier molecules. The ES bDNA assay is at least 20-fold more sensitive than the first-generation bDNA assay, yet it maintains a high level of accuracy, linearity, and reproducibility. Further, quantification values obtained with the ES bDNA assay and the first-generation bDNA assay are highly correlated, thus allowing for meaningful comparisons of HIV-1 RNA levels in specimens tested with either assay. The ES bDNA assay may be useful in determining the prognostic value of HIV-1 RNA levels of below 10,000 copies per ml and in assessing the clinical benefit of antiretroviral therapy-induced decreases in plasma HIV-1 RNA sustained at levels of below 10,000 copies per ml.
350. Among other features this flags cruciform probes as a feature which has allowed better sensitivity and consequently lower background and enhanced signal.
351. The assumption on which I am approaching Kern is that the skilled person would come to it as a result of seeing the cross-reference in Collins in relation to cruciform probes. They would read Kern carefully but be guided by that objective. What that means in detail in the context of the anticipation and obviousness attacks (especially the former, there being no material dispute about the latter) is something I consider when I come to assess the merits of those attacks.
352. The relevant discussion of probe design relied on by MI is this section in the Results part of the paper, which refers to Figure 1, parts 1A, 1B and 1C:
Principles of the ES bDNA assay. An ES bDNA assay was developed by modifying the Quantiplex HIV RNA 1.0 assay originally described by Pachl et al. (22) to reduce the background level and increase the number of bDNA amplifier molecules used to generate the signal. This was accomplished through the addition of preamplifier molecules which require specific alignment of oligonucleotide sequences and contain eight bDNA amplifier hybridization sites. Like the Quantiplex HIV RNA 1.0 assay, the ES bDNA assay uses a solution-phase sandwich assay format in which HIV-1 RNA is hybridized in solution with oligonucleotide target probes containing a unique 33-base sequence that hybridizes to a conserved region of the HIV-1 pol gene. A total of 45 target probes were designed, including 10 to mediate binding of the HIV-1 RNA to capture probes on the microwell surface (target probe set 1) and 35 to mediate binding of the HIV-1 RNA to preamplifier molecules (target probe set 2). Figure 1A illustrates the placement of the target probes by position, where each position represents a unique 33-base sequence. The first position starts at nucleotide 244 and the last position ends at nucleotide 2833 in the pol gene of model HIV strain SF2 described by Gerald Myers (Los Alamos sequence database). Each of target probes in set 1 contains a common 16-base overhang sequence that hybridizes to the capture probes on the microwell surface. As shown in Fig. 1B, the design of target probe set 2 is such that two target probes must be bound to adjacent regions of the HIV-1 RNA for efficient hybridization to the preamplifier molecule to occur. The longer sequence established by the binding of two sequential overhang sequences of target set 2 stabilizes the hybridization of the preamplifier molecule into a cruciform hybrid resembling a Holiday junction noted during DNA recombination. By design, hybridization of the preamplifier to a shorter overhang sequence alone is thermodynamically unstable. Two preamplifier molecules were designed to bridge neighboring target probes.
353. Figure 1 with its legend is as follows:
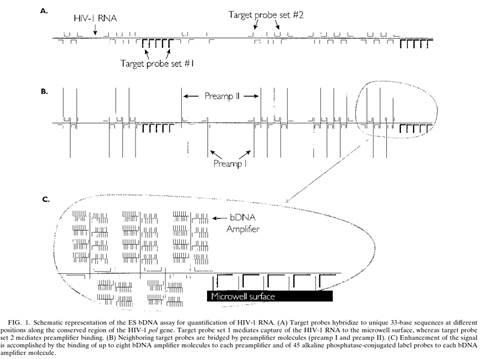
354. Figure 1C illustrates the amplification by the bDNA once the preamplifiers have been attached. It is not directly relevant to understanding what the "cruciform" arrangement is - that is set up when the long thin vertical lines "Preamp II" and "Preamp I" are added as shown in Figure 1B. The text explains that two target probes are needed "for efficient hybridisation".
355. The skilled person would understand after reading this text the sort of thing that was meant by a Holliday junction. Dr Wolf drew an illustrative arrangement of this kind in her first report:
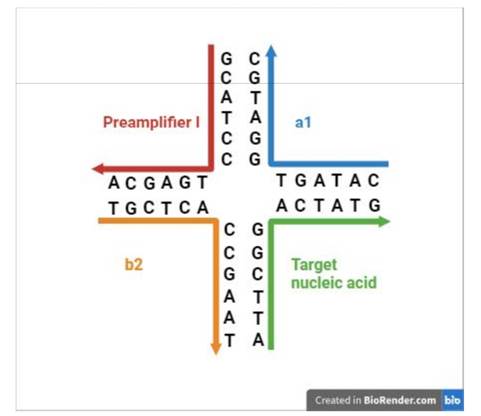

356. I provide this by way of explanation and as part of what the skilled person would work out that Kern was saying. It helps to see why the arrangement is called cruciform - although here the preamplifier and target nucleic acid are shown as L shapes, so the orientation is different from in Figure 1 - and to see that there are four double stranded arms joined together. However, I bear in mind that this is not, as such, part of the disclosure of Kern and does not enter into the anticipation analysis.
357. I will deal with Collins alone first, and then with the allegation about combining it with Kern.
358. I have dealt with the disclosure of Collins above. I find that it does disclose, most particularly in the last two sentences of the long paragraph in the right hand column of page 2983, the use of the system 8 bDNA assay in an in situ format and with a "cruciform design" (there are other options in the paper, but this one is individually described; the question is what it means).
359. However, Collins does not on its face contain a clear and unmistakable disclosure of:
i) A kit as required by feature A of claim 1 of EP439.
ii) Fixing and permeabilising the cell (feature C of claim 1 of EP572) or a reagent for permeabilising (feature B of claim 1 of EP439).
iii) The non-overlapping requirements (multiple features of both Patents).
360. MI argued that fixing and permeabilising the cell was necessarily, inevitably implicit in an in situ assay and I agree with this. It also argued that that would require a permeabilising reagent, which I also accept, and that as a result there would have at some point to be a kit. I reject that last step in the reasoning: a kit is an identifiable thing, an actual collection of components. The fact that the components would be used at some point does not make them a kit.
361. MI argued that the non-overlapping features would be implicit in the disclosure of cruciform LEs/cruciform design. I do not see how that is so to the standard necessary for anticipation, especially since I have held that the cruciform design was not CGK. There is ample in Collins to cause the skilled person to appreciate in somewhat general terms what is said - that two LEs cooperate and reduce non-specific binding - and it would incentivise them to learn more by going to Kern. That is no doubt why ACD does not and could not resist the combining for the purposes of obviousness, but Collins does not meet the standard for anticipation.
362. MI's position is better in relation to the combination of Collins and Kern as to the non-overlapping requirements (it makes no difference on the "kit" point). The twin references to Kern by means of "(18)" in the left and right hand columns on page 2983 are pointers to the skilled person that there is relevant and interesting information on the subject of the cruciform design to be found in Kern. But they do not instruct the skilled person what that information will be or how to use it.
363. So the skilled person would have to go to Kern and see what they found. I agree that they would know they should be looking for disclosure about the cruciform arrangement/LEs and I agree that with that knowledge the skilled person would work out that they should be looking at figure 1 and the associated disclosure on page 3198 of Kern. I also agree, and again this is why ACD cannot resist the combination of Collins and Kern for the purposes of obviousness, that the skilled person would fairly easily be able to work out what is going on and what they should do to achieve what is called the "cruciform hybrid resembling a Holliday junction". But all of this is a process of inference and filling in blanks going beyond what Kern itself specifically discloses.
364. I therefore reject the allegation of anticipation by the combination of Collins and Kern on the basis that the cross-reference is somewhat too general and the disclosure when one gets to Kern is not quite clear enough, two points which go together (ACD focused very much on the former).
365. ACD also argued that Collins, or Collins and Kern together, are not enabling. That must fail given my views on the CGK and anyway I am unable to see what it is that the skilled person could not deal with in implementing the teachings if I were wrong about the disclosure. I think that in substance ACD was confusing motivation with enablement. Even if it proved a lack of motivation to perform the relevant combination of Collins and Kern, that would not help it if there was a clear and unmistakable disclosure, and indeed that is the very reason why MI maintained the anticipation attacks.
366. I have dealt with the disclosure of Collins and of Kern individually above. I have also dealt with the case that in combination they anticipate the Patents, which depends on what they clearly and unambiguously disclose. I am now moving on to obviousness, where I need to consider matters to a different standard, and in particular considering what the skilled person might infer and what they would be motivated to do.
367. In this context, the key passage of Collins is still the first four paragraphs of the Discussion, most importantly the second and fourth paragraphs.
368. As I have already said, the key dispute is whether the skilled person would think that there were reasonable prospects of success for the use of the system 8 bDNA assay with cruciform probes in an in situ setting.
369. As I have also already said, this turns largely on whether there was, as a matter of CGK, a prejudice against taking an in vitro method and adapting it for in situ use. I have concluded that there was not, but that it would be expected that the task would be an empirical one which might pose some degree of practical difficulty, in respects which could not be predicted in advance but which the skilled person would expect to be able to overcome in due course.
370. As a matter of teaching, this section of Collins discloses in the first paragraph that the system 8 bDNA assay is sensitive, in large part owing to the use of isoC/isoG reducing background and improving amplification. It teaches that better sensitivity could be achieved by concentrating larger samples. Pausing there, the teaching is that sensitivity is good but could usefully be improved still further, and this would interest the skilled person and motivate them to consider the matter further.
371. The second paragraph goes on to say that increasing volume is one approach but that in addition the reduction of background by reducing NSB would be desirable. It cross refers to Kern and that is where the skilled person would look for more details of cruciform probes, which Collins suggests as a way to get rid of background. The authors are not saying that that will definitely provide the improvement discussed (only "in theory"), but they are saying that the objective would be to improve sensitivity. So again and for the same reasons a motivation is provided.
372. The fourth paragraph refers to the potential use of the system 8 bDNA assay in situ (along with in filter assays). The authors explain the problem of non specific binding in in situ assays.
373. There is an intervening reference to in situ PCR being the standard for detection of single copy DNA sequences in cells. As a matter of CGK the skilled reader would understand that that was no longer current because the problems with in situ PCR had come to be understood widely (and a then-existing problem with mRNA detection is also mentioned in Collins), but this does not undermine the logic of the improved set-ups the authors are discussing.
374. The authors also say that quantification in situ may be possible. They go on to repeat the possibility to use cruciform probes to get the best specificity (last sentence, the context still clearly being in situ).
375. These paragraphs therefore provide a motivation for using cruciform probes along with the system 8 bDNA assay (better sensitivity) and has a rationale for the application to in situ assays.
376. Dr Wolf and ACD pointed out that these statements are made in the discussion section, and that that is the part of scientific papers where relatively more speculative proposals are made. I agree with that so far as it goes, and will take it into account, but it does not add much if anything in the light of my finding that there was no general prejudice against taking in vitro techniques forward to in situ use, and in the absence of any concrete reason why cruciform probes should be a problem. It also does not undermine the significant positive motivation to try.
377. I have rejected the anticipation attack by which MI asserted that Collins with its cross-reference to Kern clearly and unambiguously disclosed the use of bDNA with cruciform probes in situ to the relevant legal standard.
378. But ACD did not oppose the proposition that the two documents would be obvious to read together in such a way that the skilled person would think of that combination of features.
379. The Pozzoli analysis is not of ready application to this case, or at least does not provide much benefit, because all the features of the claim would occur to the skilled person, and the issue is prospects of success.
380. I have identified the skilled person and the CGK above. I do not feel I need to paraphrase the inventive concept separately from the claims for this purpose.
381. Additionally, I find that the skilled person would readily understand the benefits that the combination would bring if done successfully: improved specificity while being able to perform the kinds of studies to which ISH is directed. This is clearly flagged in the prior art.
382. So the nub of the question on obviousness is simply whether there was a reasonable expectation of success.
383. ACD did not put forward any reason specific to Kern, Collins or the combination of both why the skilled person would positively think that prospects of success were lacking. Its case was almost entirely founded on its mindset argument that I have covered above in connection with the topics of the skilled person and the CGK (I say "almost" because it also relied on secondary evidence which I address in a moment).
384. I have rejected the mindset argument and subject to the secondary evidence issue that is effectively the end of ACD's opposition to a finding of obviousness given the disclosure of Collins with Kern and the motivation to try.
385. I have, second, also found that in any event Player was CGK and that too is fatal, because if, contrary to my primary finding, the skilled person did have any doubts about whether the proposal in Collins and Kern was worth trying because of uncertainty over using bDNA in situ, those doubts would be allayed by knowledge, from CGK that it had been done by Player.
386. Third, if the skilled person had any doubts over prospects of success and did not know of Player by way of CGK, I hold that they would not just give up on an otherwise appealing idea but would, following an obvious course in seeking to understand whether or not their doubts had substance, find Player on a routine literature search which they would undertake, the better to assess the prospects of success.
387. I do not overlook and have had in mind that there are other routes that the skilled person could follow from Kern, although this was not at all the focus of ACD's argument. I do not think it makes any material difference; bDNA with cruciform probes in situ is a possibility which is quite prominent (just not with the high level of clarity needed for anticipation) and has obvious attractions. I also do not overlook ACD's arguments that the skilled person might think that more demanding applications of what Collins discloses (for example, quantification) might be appreciably more difficult, but that would not stand in the way of using it for more straightforward tasks such as detection of low abundance RNAs, and anyway the claims of the Patents are not limited to quantification etc. Finally, while I have said that ACD did not put forward positive reasons to think that the approach would not work, it did point to some matters which it said would give rise to uncertainty. For example, Dr Wolf said that the bDNA "tree" might be sterically (i.e. spatially) hindered in the context of in situ use in a way which had not been observed or was not relevant in vitro. I found this all speculative and such concerns would be allayed by the fact that Player had succeeded, and could in any event go no further than providing some possible reason why success was not guaranteed. I have not made my decision of obviousness on the basis that it was guaranteed, but on the basis that the prospects of success would be assessed by the skilled person as not only reasonable, but good.
388. I will address this only very briefly since ACD accepts that if cruciform probes were CGK then the Patents are obvious over Player.
389. Player is a 2001 publication from Bayer; Bayer had by then taken over Chiron. Its title is "Single-copy Gene Detection Using Branched DNA (bDNA) In Situ Hybridization". The technique is tested using HPV DNA in whole cells in a setup where it could be known that there were only very few copies of the DNA being tested for.
390. The experimental set-up is shown in figure 1:
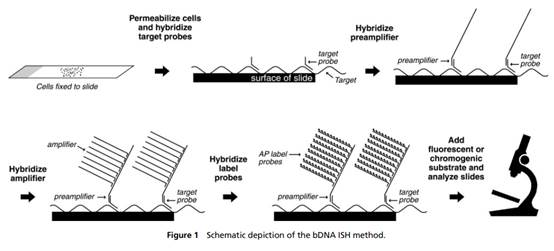
391. This shows the now-familiar frond-like arrangement of the bDNA amplification system. It is explicitly in situ. But its target probes are not cruciform and it therefore lacks the features of the claims requiring multiple capture probes and the non-overlapping features (I paraphrase).
392. I can be very short about this. For the Patents to be obvious over Player it would be necessary for it to be obvious to enhance what it teaches by the use of cruciform probes - that is the difference for the purposes of Pozzoli.
393. There is however no disclosure in Player of the use of cruciform probes, or any pointer to it. MI's argument to meet this was that cruciform probes were CGK but I have rejected that. MI also faintly argued that a skilled person wanting to progress Player would find out about cruciform probes by a literature search, but I considered and rejected that argument when addressing CGK.
394. So the attack over Player fails.
395. ACD argued that the Chiron/Bayer group had all the necessary pieces of the puzzle to make the invention of the Patents, in particular in that they used bDNA with cruciform probes, but did not successfully put it into practice in an in situ assay, even though they clearly had the idea, given that they said so in Collins.
396. ACD also put this in terms of what the skilled person would think at the Priority Date reading Collins: they would know (at least if they looked into it, as I agree they would if considering pursuing it) that despite Collins making obvious the proposal of bDNA with cruciform probes in situ in 1997, Chiron had not done it.
397. I agree that Chiron had all the information necessary, but what is entirely lacking is evidence, or reason to think, that they tried and failed, or that they were deterred from trying by the perceived difficulty.
398. Chiron was a major player in this field, but that does not mean that it had infinite resources. It can be seen from the public materials that it had major business and scientific imperatives with HIV and HCV assays; it may have prioritised those. It is possible that its in situ bDNA approach had enough sensitivity for its purposes without cruciform probes. It is possible that it tried with cruciform probes and did not publish. It cannot be ruled out that indeed it tried and failed, but even if I assumed that, which I am not willing to do, it might be because the effort was directed to a very challenging particular implementation, or they might have been unlucky. The sale to Bayer may also have had an effect.
399. There are so many possibilities that this cannot help ACD. I also note that ACD has had the assistance of Dr Urdea in actual or potential proceedings in the USA; Counsel for ACD accepted that that was so on the basis of a remark in Dr Wolf's evidence. So if there had been real issues at Chiron, ACD had the means to seek to lead evidence about them. There are reasons why it might not wish to do so even if there were such difficulties, for example because it wanted to preserve Dr Urdea's ability to give evidence in the USA in due course without his having been exposed to challenge here. Whilst I therefore cannot be certain of the position, I think the situation is plain enough that I can draw the inference that had there been a really convincing history about Chiron's work, ACD would have told it. At the very least, it is a material factor against ACD on this point.
400. Related to this point, ACD relied on the time that it appears to have taken Chiron to try bDNA in situ from when it had the initial idea. The problem with this is that there is no way to know when it actually started work or when it finished. So I give this no weight, either.
401. ACD also relied on the fact that Dr Wolf gave evidence that when asked about ACD's intention to pursue the approach described in the Patents at the time, she expressed scepticism. I do not doubt that her account reflected her recollection, but it was apparent that her recollection was, understandably, not complete. More significantly, it emerged in the cross-examination that her scepticism was not about whether bDNA would work at all in situ, but whether ACD's specific proposed uses, which were ambitious, could be achieved.
402. Thus I do not think there is any help to be had for ACD in the secondary evidence. Even if there had been, it is called secondary evidence for a reason and the primary evidence is really overwhelming in this case.
403. At the start of the trial MI had a "free-standing" (i.e. not a squeeze) insufficiency argument that the inventions of the Patents could only work at appropriate melting temperatures but that the claims were not limited to those temperatures. The argument was dropped in MI's written closing submissions.
404. Two insufficiencies were maintained as squeezes (I take their formulation from MI's closing written submissions, paragraph 231):
a. That if ACD say (as they do) that it would not be obvious to transfer the in vitro assay of the prior art to an in situ format because there would be no expectation of success, then the Patents are insufficient because they do not demonstrate that the claimed methods/kits work.
b. That if ACD contend that a successful ISH assay requires a careful balancing of multiple parameters in order to be successful (sample preparation conditions, probe size, hybridization conditions etc.), then the claimed methods are insufficient because it is not plausible that they will work across their breadth. The claims are very wide and for all practical purposes unlimited in relation to the parameters that are said to require careful balancing.
405. Since I have held the Patents obvious the insufficiencies essentially do not arise but I will sweep up some minor points.
406. First, I note that ACD sought to break up the first squeeze by saying that the Patent refers to Kenny and Player. I have held that Player was CGK and that anyway the skilled person would find it by a routine literature search in the relevant scenario. So ACD's way out of the squeeze does not help it.
407. Second, ACD hinted at a reliance on the fact that Player only shows success of bDNA in situ for a single label extender, not two. That cannot work to break up the squeeze because if it caused the skilled person to lose confidence in the prior art the same would apply to the Patents. Also, I do not consider there was any reason to think that using two label extenders instead of one would cause a problem.
408. Third, ACD through Dr Wolf argued that Player does not prove that in situ bDNA was clearly better than other alternatives. If accepted this would tend to negate the Patents providing any benefit, too, but anyway on my reasoning the skilled person's motivation would lie in the greater specificity taught by Collins and Kern and would only be looking to Player for additional confidence that bDNA would work in situ (if they needed any such additional confidence, which I do not think they would).
409. As to the second squeeze, my findings on obviousness are that the skilled person would have sufficient confidence that they could find workable sets of conditions such that they would have the necessary expectation of success taking Collins and Kern forwards. I have not found that they would expect all sets of conditions to work; they would think that some would not. I do not think this would amount to an insufficiency, if it arose. That is because, very briefly, the Patents' inventions are not about choosing individual sets of detailed conditions so they are not "relevant ranges" in the sense explained by Birss J in Illumina (supra). However, since my factual findings and decision on obviousness make all of this irrelevant to the result, I will say no more about it.
410. EP439 was opposed in the EPO by Affymetrix Inc.
411. The Opposition Division ("OD") rejected the opposition and I was provided with its reasons, dated 2 January 2017. Those followed an oral hearing in December 2016.
412. All four of the prior art citations relied on by MI at this trial were considered by the OD.
413. As the decision records, Affymetrix had withdrawn its opposition by the date of the oral proceedings. The fact that it did not argue its case at the hearing reduces the weight that I can give the decision.
414. In addition, it appears that the arguments are likely to have been materially different from those which I heard, and the evidence was quite plainly not the same.
415. I note that I have reached the same conclusion on anticipation as did the OD, although for reasons which are somewhat different. I have reached a different conclusion on obviousness over Collins (see 45.3.2 in the decision) but the OD provided minimal reasoning for its conclusions, understandably in the circumstances and in view of the fact that Urdea and Kern seem to have been more of a focus.
416. Given that the arguments and evidence were so different the OD decision does not cause me to question my conclusions.
417. My conclusions are:
i) The Patents are invalid for obviousness over Collins with Kern.
ii) The anticipation attacks over Collins and over Collins with Kern fail.
iii) The obviousness attack over Player fails.
iv) It is not necessary to decide validity over Urdea.
v) Insufficiency was only run as a squeeze and since the Patents are obvious, it does not arise for separate decision.
vi) The added matter squeeze does not arise in the light of the way that ACD ran its infringement case.
vii) Had the Patents been valid, EP572 but not EP439 would have been infringed by the acts to be considered at this trial.
418. I will hear Counsel as to the form of Order if it cannot be agreed. I direct that time for seeking permission to appeal shall not run until after the hearing on the form of Order (or the making of such Order if it is agreed). I draw attention to paragraph 19.1 of the Patents Court Guide, which says that a hearing on the form of Order should take place within 28 days of hand down. In the present case, 28 days from hand down will be 21 May 2024.
